Taille et part du marché du traitement des maladies lysosomales de surcharge
VUE D’ENSEMBLE DU MARCHÉ
| Période d'étude | 2022 - 2031 |
|---|---|
| Taille du Marché (2026) | 5.12 Milliards de dollars |
| Taille du Marché (2031) | 7.39 Milliards de dollars |
| Taux de croissance (2026 - 2031) | 7.65% CAGR |
| Marché à la Croissance la Plus Rapide | Asie-Pacifique |
| Plus Grand Marché | Amérique du Nord |
| Concentration du Marché | Moyen |
Acteurs majeurs *Avis de non-responsabilité : les principaux acteurs sont triés sans ordre particulier Image © Mordor Intelligence. La réutilisation nécessite une attribution sous CC BY 4.0. | |
Analyse du marché du traitement des maladies lysosomales de surcharge par Mordor Intelligence
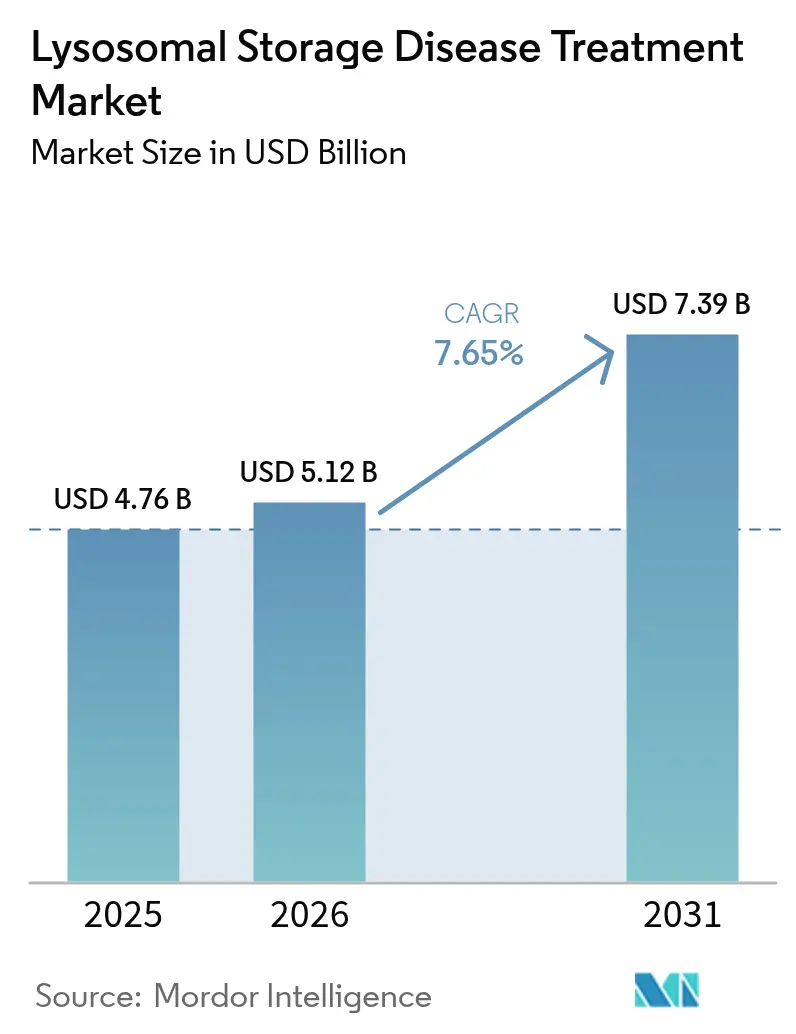
La taille du marché du traitement des maladies lysosomales de surcharge était évaluée à 4,76 milliards USD en 2025 et devrait croître de 5,12 milliards USD en 2026 pour atteindre 7,39 milliards USD d'ici 2031, à un TCAC de 7,65 % au cours de la période de prévision (2026-2031). L'accélération des approbations pour les thérapies géniques à dose unique, l'élargissement des indications pour la thérapie enzymatique substitutive et les programmes de dépistage génomique néonatal ont déplacé l'initiation du traitement vers des stades présymptomatiques, élargissant ainsi la population adressable. L'intensité concurrentielle s'accroît alors que les acteurs historiques de la thérapie enzymatique font face à des challengers développant des vecteurs pénétrant la barrière hémato-encéphalique et des substrats oraux à petites molécules. L'augmentation à l'échelle industrielle de la fabrication de vecteurs viraux reste un goulet d'étranglement, même si des capitaux affluent vers les organisations de développement sous contrat. La croissance parallèle des services de perfusion à domicile, qui réduisent les coûts d'administration de 25 à 50 %, remodèle les modèles de prestation de soins.
Principaux enseignements du rapport
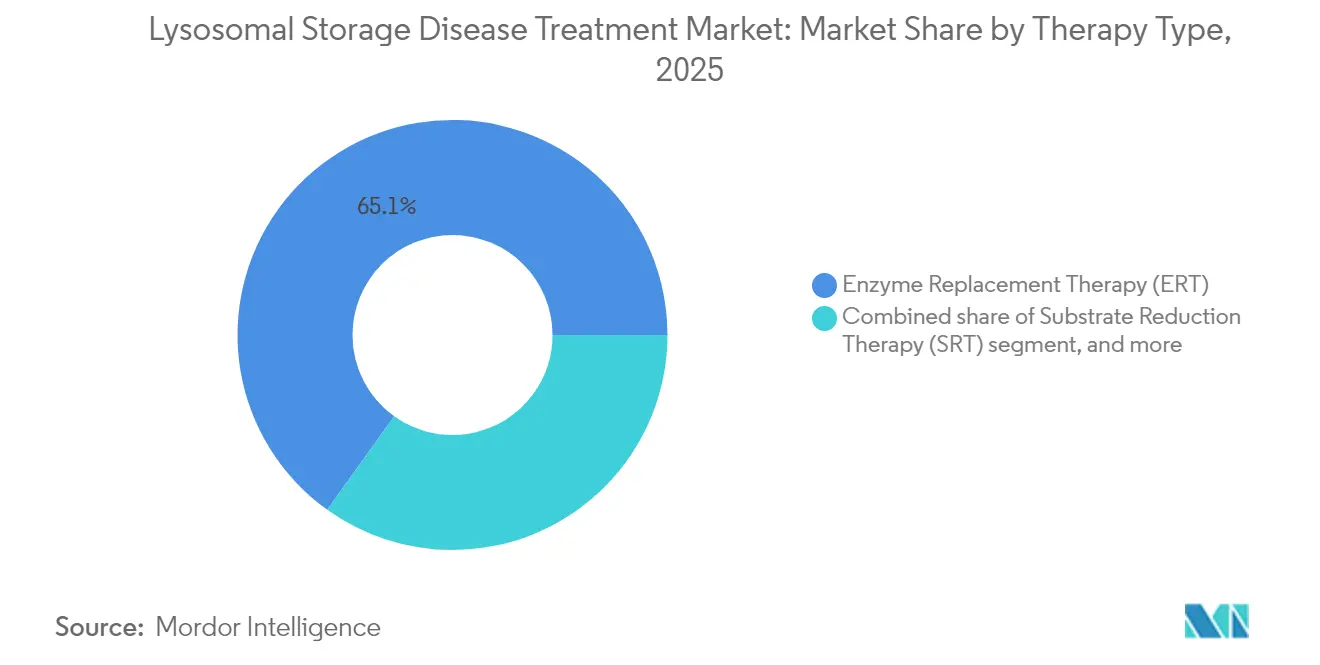
- Par type de thérapie, la thérapie enzymatique substitutive a représenté 65,10 % de la part du marché du traitement des maladies lysosomales de surcharge en 2025 ; la thérapie génique devrait progresser à un TCAC de 10,22 % jusqu'en 2031.
- Par modalité, les biothérapies intraveineuses représentaient 55,20 % de la taille du marché du traitement des maladies lysosomales de surcharge en 2025, tandis que la thérapie orale à petites molécules devrait croître à un TCAC de 10,12 % jusqu'en 2031.
- Par type de maladie, la maladie de Gaucher représentait 28,10 % de la taille du marché du traitement des maladies lysosomales de surcharge en 2025 ; la maladie de Niemann-Pick progresse à un TCAC de 10,35 % jusqu'en 2031.
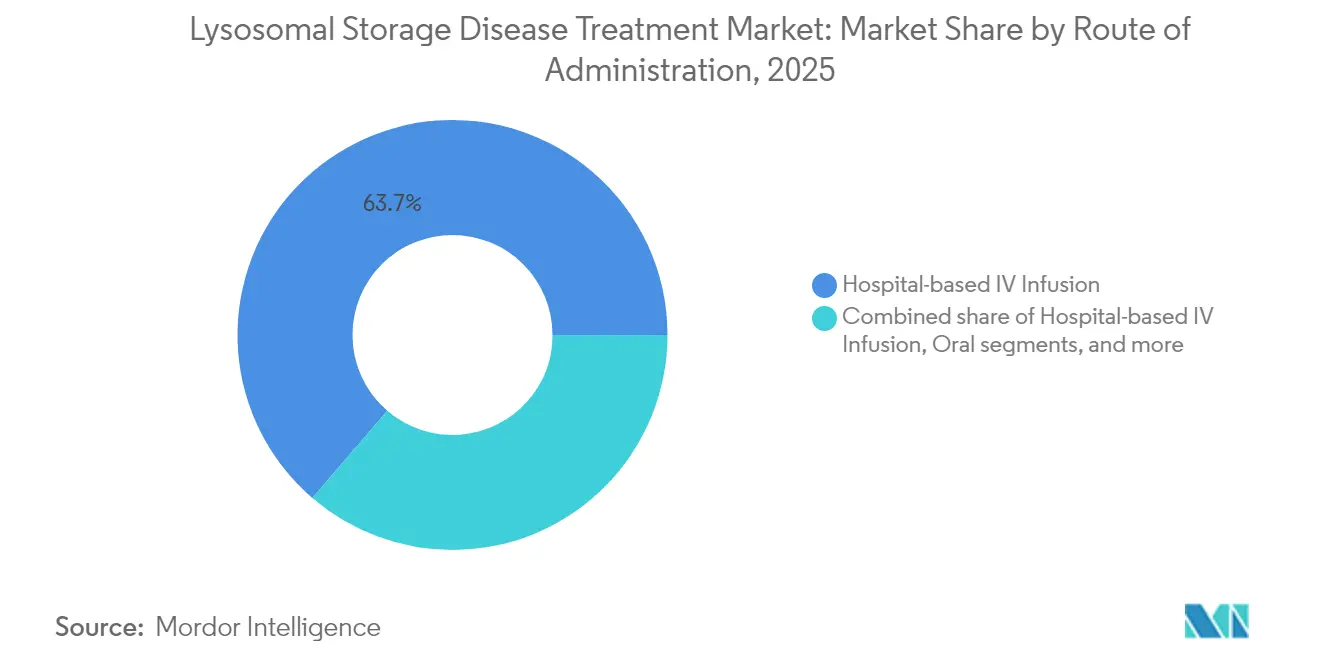
- Par voie d'administration, la perfusion IV en milieu hospitalier représentait 63,70 % de la part du marché du traitement des maladies lysosomales de surcharge en 2025, tandis que la perfusion IV à domicile devrait afficher un TCAC de 11,05 % jusqu'en 2031.
- Par utilisateur final, les hôpitaux tertiaires ont capté 65,60 % des revenus en 2025, tandis que les environnements de soins à domicile devraient croître à un TCAC de 11,20 % jusqu'en 2031.
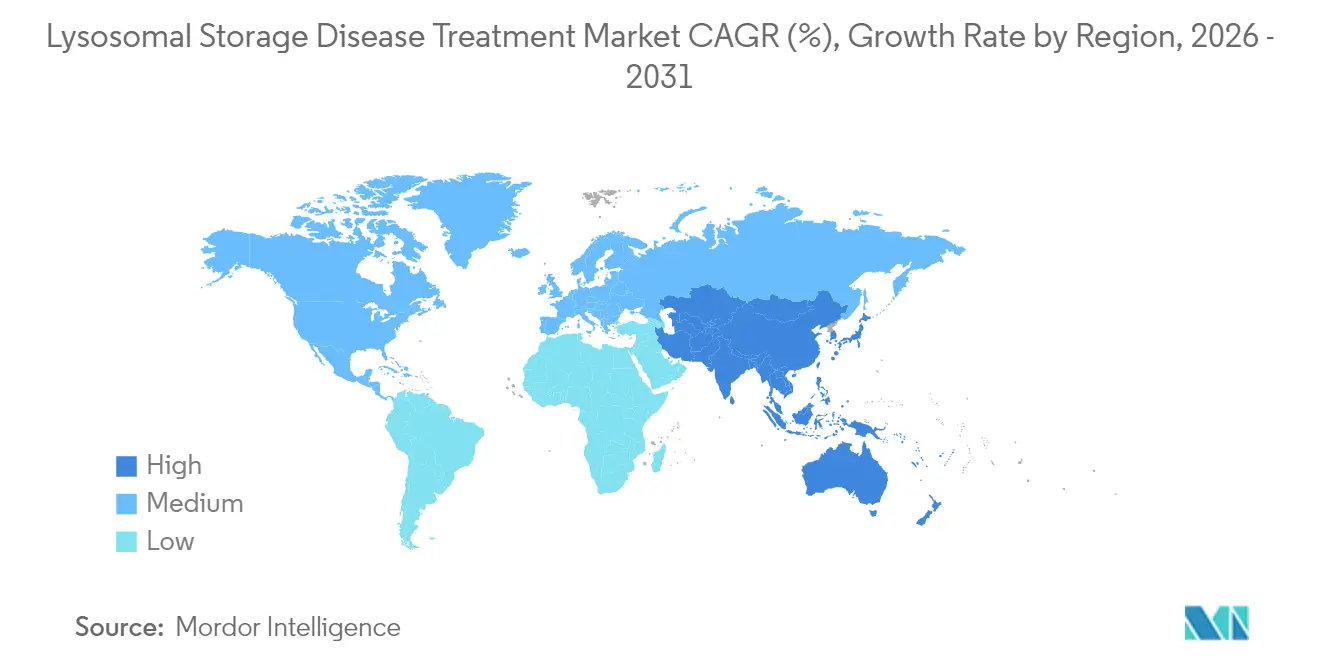
- Par géographie, l'Amérique du Nord a capté 42,10 % des revenus en 2025, tandis que la région Asie-Pacifique devrait croître à un TCAC de 9,15 % jusqu'en 2031.
Remarque : Les chiffres de la taille du marché et des prévisions de ce rapport sont générés à l’aide du cadre d’estimation propriétaire de Mordor Intelligence, mis à jour avec les données et analyses les plus récentes disponibles en 2026.
Tendances et perspectives mondiales du marché du traitement des maladies lysosomales de surcharge
Analyse de l'impact des moteurs*
| Moteur | % d'impact sur les prévisions du TCAC | Pertinence géographique | Calendrier d'impact |
|---|---|---|---|
| Prévalence mondiale croissante des maladies lysosomales de surcharge | +1.2% | Mondial – détection la plus élevée sur les marchés développés | Moyen terme (2 à 4 ans) |
| Augmentation des désignations de médicaments orphelins et des incitations | +1.0% | Leadership de l'Amérique du Nord et de l'UE ; adoption s'étendant à l'échelle mondiale | Court terme (≤ 2 ans) |
| Avancées dans les technologies de diagnostic et le dépistage précoce | +1.8% | Amérique du Nord et UE en tête ; déploiement rapide en Asie-Pacifique | Court terme (≤ 2 ans) |
| Expansion des approbations de produits de thérapie enzymatique substitutive | +1.3% | Mondial, plus fort en Amérique du Nord et dans l'UE | Moyen terme (2 à 4 ans) |
| Thérapies géniques et cellulaires émergentes pour les maladies lysosomales de surcharge neuropathiques | +2.1% | Mondial, concentré dans les grands pôles pharmaceutiques | Long terme (≥ 4 ans) |
| Collaborations stratégiques et afflux d'investissements dans les maladies rares | +1.5% | Noyau en Amérique du Nord et dans l'UE ; expansion vers les marchés émergents | Moyen terme (2 à 4 ans) |
| Source: Mordor Intelligence | |||
Prévalence mondiale croissante des maladies lysosomales de surcharge
Le dépistage génomique néonatal à Shanghai a identifié une incidence des maladies lysosomales de surcharge de 1 pour 1 856 naissances vivantes, nettement supérieure aux estimations historiques et confirmant la valeur du dépistage proactif. Le programme CARE de l'Administration nationale des produits médicaux de Chine encourage le dépôt de demandes de médicaments orphelins et a augmenté les soumissions de nouveaux médicaments expérimentaux de 32 % depuis 2024. Des hausses similaires de prévalence apparaissent dans le registre national de la Corée du Sud, soulignant le passage d'un diagnostic anecdotique à un dépistage à l'échelle de la population. À mesure que les registres s'élargissent, la demande d'intervention précoce augmente, réduisant le délai diagnostique de six ans à moins de six semaines dans les principaux centres tertiaires. Les pipelines thérapeutiques sont donc recalibrés vers les indications à début infantile où le bénéfice clinique est le plus prononcé.
Avancées dans les technologies de diagnostic et le dépistage précoce
La spectrométrie de masse en tandem de deuxième génération permet désormais de dépister jusqu'à huit maladies lysosomales de surcharge en un seul test avec moins de 0,5 % de faux positifs, surpassant les panels d'activité enzymatique. L'intégration du séquençage de nouvelle génération confirme les variants pathogènes et guide la thérapie de précision, comme la sélection du migalastat pour les patients atteints de la maladie de Fabry présentant des mutations accessibles. Les délais d'obtention des résultats pour le génotypage confirmatoire sont passés en dessous de sept jours, permettant l'initiation du traitement pendant les séjours en unité de soins intensifs néonatals. L'adoption par les systèmes de santé est la plus rapide au Canada et en Allemagne, où le remboursement gouvernemental couvre à la fois les tests biochimiques et génomiques. Le diagnostic précoce catalyse l'acceptation par les payeurs des thérapies géniques à haute valeur en démontrant les compensations à long terme des coûts évités liés aux handicaps.
Thérapies géniques et cellulaires émergentes pour les maladies lysosomales de surcharge neuropathiques
La thérapie par cellules souches autologues à dose unique Lenmeldy a permis le maintien des étapes motrices chez 90 % des patients atteints de leucodystrophie métachromatique traités, contre 30 % historiquement observés dans les cohortes non traitées[1]Agence américaine des produits alimentaires et médicamenteux (U.S. FDA), « Programme de bons de révision prioritaire pour les maladies pédiatriques rares », fda.gov. RGX-121 a réduit le sulfate d'héparane dans le liquide céphalorachidien de 85 % et a permis à 80 % des patients d'interrompre les perfusions hebdomadaires. Les capside AAV9 pénètrent la barrière hémato-encéphalique plus efficacement que les vecteurs antérieurs, maintenant l'expression enzymatique pendant au moins 36 mois dans les essais portant sur la maladie de Gaucher et la gangliosidose GM1. Les rendements de fabrication, autrefois limités à 1e15 génomes vectoriels par lot, ont doublé grâce aux technologies HEK293 en suspension, réduisant le coût des marchandises de 38 %. Les agences de réglementation instituent des mécanismes d'examen continu qui ont réduit les délais d'approbation de neuf mois en moyenne, intensifiant la vélocité concurrentielle.
Collaborations stratégiques et afflux d'investissements dans les maladies rares
L'accord de licence de 110 millions USD conclu par REGENXBIO avec Nippon Shinyaku pour les programmes MPS illustre un pivot vers des structures de co-commercialisation régionales qui répartissent les risques et accélèrent l'entrée sur le marché. L'acquisition par BioMarin d'Inozyme pour 270 millions USD sécurise une thérapie enzymatique substitutive en phase avancée pour la déficience en ENPP1, consolidant la profondeur transversale du portefeuille. Le financement par capital-risque reste résilient, Glycomine ayant levé 115 millions USD en Série C pour faire avancer les thérapeutiques liées aux troubles de la glycosylation. Les partenariats se concentrent désormais sur les plateformes navettes pour la barrière hémato-encéphalique et l'ingénierie modulaire des capsides, des domaines qui devraient attirer plus de 2 milliards USD de nouveaux capitaux d'ici 2027. Ces alliances compriment les délais de recherche et développement tout en donnant aux petites biotechnologies accès aux capacités de fabrication et à l'expertise réglementaire établies.
Analyse de l'impact des contraintes*
| Analyse de l'impact des contraintes | (~) % d'impact sur les prévisions du TCAC | Pertinence géographique | Calendrier d'impact |
|---|---|---|---|
| Coûts élevés des traitements et défis du remboursement | −1.8% | Mondial – le plus aigu sur les marchés émergents | Long terme (≥ 4 ans) |
| Pénétration limitée de la barrière hémato-encéphalique par les thérapies actuelles | −1.2% | Mondial – impacte les formes neuronopathiques | Moyen terme (2 à 4 ans) |
| Évolutivité de la fabrication et contraintes d'approvisionnement | −1.4% | Mondial, centré sur les pôles de production de thérapies géniques | Moyen terme (2 à 4 ans) |
| Préoccupations à long terme concernant la sécurité et l'immunogénicité | −1.0% | Mondial, affectant toutes les modalités avancées | Long terme (≥ 4 ans) |
| Source: Mordor Intelligence | |||
Coûts élevés des traitements et défis du remboursement
Lenmeldy est commercialisé à 4,25 millions USD, ce qui en fait la thérapie la plus chère au monde et contraint les payeurs à adopter des contrats basés sur les résultats liant le remboursement aux paramètres de la fonction motrice[2]Thomas Mueller et al., « Charge financière de la thérapie enzymatique substitutive en Allemagne », Orphanet Journal of Rare Diseases, ojrd.biomedcentral.com. En Allemagne, les dépenses annuelles moyennes de thérapie enzymatique substitutive pour la maladie de Fabry s'élèvent à 369 047 EUR (405 952 USD) par patient, dont 94 % correspondent au coût d'acquisition du médicament. Les programmes Medicaid excluent les dispositifs de thérapie génique des paiements par capitation pour atténuer les chocs financiers ponctuels. Les assureurs privés limitent la couverture aux cas génétiquement confirmés et exigent des données longitudinales sur les biomarqueurs, prolongeant l'accès dans 17 États américains. Les modèles de paiement innovants de type annuité progressent lentement en raison des directives réglementaires non résolues concernant l'amortissement pluriannuel.
Pénétration limitée de la barrière hémato-encéphalique par les thérapies actuelles
L'imiglucérase intraveineuse ne franchit pas la barrière hémato-encéphalique, laissant la maladie de Gaucher neuronopathique non prise en charge malgré le contrôle des symptômes somatiques. La transcytose médiée par récepteurs utilisant des ligands de transferrine a démontré une absorption enzymatique quadruplée dans des modèles précliniques de la maladie de Pompe, mais attend encore une confirmation en phase 2. La cerliponnase alfa intrathécale prolonge la survie des patients atteints de CLN2, mais nécessite un accès trimestriel par cathéter neurochirurgical, limitant son adoption à grande échelle. Les vecteurs géniques présentent un tropisme supérieur pour le système nerveux central, bien que les anticorps neutralisants contre l'AAV9 excluent jusqu'à 25 % des adultes de l'inscription aux essais. Les contraintes de fabrication pour les vecteurs à titre élevé persistent malgré les lignées productrices à double plasmide qui augmentent le rendement de 30 %.
*Nos prévisions considèrent les impacts des moteurs et des contraintes comme directionnels et non additifs. Les prévisions d'impact reflètent la croissance de référence, les effets de composition et les interactions entre variables.
Analyse des segments
Par type de thérapie : leadership soutenu de la thérapie enzymatique substitutive face à une montée rapide de la thérapie génique
La taille du marché du traitement des maladies lysosomales de surcharge pour la thérapie enzymatique substitutive s'élevait à 3,1 milliards USD en 2025, soit 65,10 % du chiffre d'affaires total, et demeure essentielle pour la prise en charge de la maladie de Gaucher, de la maladie de Fabry et de la maladie de Pompe. Le positionnement concurrentiel se concentre sur les enzymes de deuxième génération telles que l'avalglucosidase alfa, qui offre une absorption musculaire plus élevée que l'alglucosidase prédécesseur. Cependant, le TCAC de 10,22 % de la thérapie génique reflète sa proposition de valeur à dose unique. L'approbation de Lenmeldy illustre le bénéfice neurologique durable, tandis qu'UX111 d'Ultragenyx est en tête du calendrier d'examen FDA 2025 pour le syndrome de Sanfilippo. La réduction du substrat et les chaperons pharmacologiques apportent des gains incrementaux pour les cohortes spécifiques aux mutations, renforçant les trajectoires de médecine de précision au sein du marché du traitement des maladies lysosomales de surcharge. La convergence du pipeline vers des schémas thérapeutiques combinés, tels que Pombiliti plus Opfolda, vise à amplifier le trafic intracellulaire et à prolonger les intervalles de traitement.



Par modalité : les schémas oraux gagnent du terrain face aux normes parentérales
Les biothérapies intraveineuses ont conservé 55,20 % des revenus en 2025, ancrées par les perfusions hebdomadaires d'imiglucérase, d'agalsidase bêta et d'alglucosidase alfa. Les réducteurs de substrat oraux à petites molécules — venglustat, lucerastat et Cerdelga approuvé — enregistrent la croissance la plus élevée à un TCAC de 10,12 %, portés par les avantages en termes d'adhérence et l'absence de réactions aux perfusions. Le mix des modalités se rééquilibre davantage à mesure que le chaperon oral migalastat s'étend à d'autres génotypes de la maladie de Fabry. Les administrations intrathécales et intracérébroventriculaires sont réservées aux syndromes neuronopathiques ; des dispositifs tels que SmartFlow Neuro permettent un placement précis de la canule pour le transfert de gène Kebilidi. Les technologies de PEGylation et de fusion Fc prolongent la demi-vie des thérapies enzymatiques substitutives comme ELFABRIO, réduisant la fréquence des perfusions à un rythme mensuel. La posologie centrée sur le patient s'aligne sur l'expansion de la perfusion à domicile et souligne l'accent mis par les payeurs sur la réduction du coût total des soins au sein du marché du traitement des maladies lysosomales de surcharge.
Par type de maladie : portefeuille établi dans la maladie de Gaucher face à la dynamique rapide de la maladie de Niemann-Pick
La maladie de Gaucher reste le plus grand sous-marché, représentant 28,10 % des revenus de 2025, soutenue par trois thérapies enzymatiques substitutives commerciales et un agent oral qui diversifient la pression sur les prix. La concurrence favorise des remises périodiques sur les prix et des programmes d'accès à titre compassionnel élargis. La maladie de Niemann-Pick illustre la future cohorte à forte croissance avec un TCAC de 10,35 % jusqu'en 2031 après la double approbation par la FDA de Miplyffa (oral) et AQNEURSA (IV), mettant fin à des décennies de vide thérapeutique. La maladie de Fabry et la maladie de Pompe maintiennent une expansion à un chiffre moyen, grâce à l'adoption de la thérapie par chaperons et aux enzymes de nouvelle génération montrant des paramètres rénaux et pulmonaires supérieurs. Les mucopolysaccharidoses stimulent l'innovation dans le domaine de la thérapie génique ; RGX-111 pour la MPS IIB pourrait apporter une correction durable du système nerveux central, remettant en cause la thérapie enzymatique substitutive intrathécale chronique. Les pipelines émergents dans la gangliosidose GM1 et l'alpha-mannosidose illustrent le potentiel des espaces vierges à mesure que les données en phase précoce montrent une réduction de plus de 70 % des substrats de stockage.
Par voie d'administration : la perfusion à domicile redessine les parcours de soins
Les centres de perfusion IV en milieu hospitalier ont dispensé 63,70 % des doses en 2025, mais l'orientation des payeurs vers des sites de soins moins coûteux accélère le transfert vers les environnements à domicile, affichant désormais un TCAC de 11,05 %. Les données probantes du monde réel provenant des cohortes allemandes atteintes de la maladie de Fabry montrent une parité dans les taux d'événements indésirables entre les perfusions à domicile et en clinique, tout en réduisant les coûts hors médicaments de 42 %. Les schémas oraux poussent davantage la décentralisation, permettant l'auto-administration et réduisant les contraintes de déplacement pour les familles rurales. Les voies intrathécales, malgré les exigences chirurgicales, sont de plus en plus proposées dans des centres de neurochirurgie satellites pour élargir l'accès. Les thérapies géniques, administrées une seule fois dans des hôpitaux spécialisés, pourraient par la suite rendre superflus les besoins chroniques en perfusion, accentuant l'impact perturbateur sur l'infrastructure de prestation au sein du marché du traitement des maladies lysosomales de surcharge.
Par utilisateur final : les centres tertiaires perdent leur exclusivité au profit des soins communautaires
Les hôpitaux tertiaires gèrent encore 65,60 % des patients traités compte tenu de la complexité du diagnostic et des besoins de surveillance. Pourtant, les programmes de soins à domicile, renforcés par la télémédecine, affichent un TCAC de 11,20 % à mesure que les équipes de perfusion dirigées par des infirmières élargissent leur couverture géographique. Les cliniques spécialisées dans les maladies rares, souvent affiliées à des universités, préservent leur pertinence grâce à l'accès aux protocoles expérimentaux et à l'expertise multidisciplinaire. La posologie unique de la thérapie génique comprime les flux de revenus hospitaliers à long terme, incitant les établissements à rechercher des contrats basés sur la valeur et des services complémentaires de conseil génomique. Les payeurs pilotent des forfaits de soins intégrés qui remboursent les tests génétiques, le conseil et la prestation médicamenteuse sous un code unique, réalignant les incitations vers la gestion communautaire au sein du marché du traitement des maladies lysosomales de surcharge.
Analyse géographique
L'Amérique du Nord a représenté 42,10 % des revenus de 2025, portée par le programme de bons de révision prioritaire pour les maladies pédiatriques rares de l'Agence américaine des produits alimentaires et médicamenteux (FDA) et une couverture d'assurance commerciale étendue. Plus de 20 essais actifs de thérapie génique recrutent à travers les États-Unis, consolidant la domination de la région en matière d'innovation. La disparité au niveau des États persiste : la Californie couvre les frais de voyage et d'hébergement pour les bénéficiaires de Lenmeldy venant d'autres États, tandis que six États du Midwest limitent les thérapies géniques aux exclusions Medicaid, allongeant les délais d'accès. Le système universel canadien rembourse cinq thérapies enzymatiques substitutives sur une liste de médicaments orphelins désignés, bien que le délai entre l'approbation et le financement atteigne en moyenne 14 mois.
L'Asie-Pacifique est le territoire à la croissance la plus rapide avec un TCAC de 9,15 %. La liste actualisée des maladies rares de la Chine, qui compte désormais 207 affections, a réduit le temps d'examen réglementaire pour les agents importés à 9 mois, contre 24 mois avant 2023. Le programme de dépistage génomique pilote de Shanghai a révélé une incidence des maladies lysosomales de surcharge bien supérieure aux moyennes mondiales, ce qui a conduit à l'octroi de subventions nationales pour le déploiement à grande échelle. Le Japon s'appuie sur sa procédure accélérée Sakigake pour attirer les dépôts de biotechnologies étrangères, tandis que l'Assurance maladie nationale de la Corée du Sud couvre plus de 90 % des traitements de la maladie de Fabry, démontrant une maturité avancée des payeurs. La politique indienne sur les maladies rares subventionne jusqu'à 2 millions INR (2,2 millions USD) par patient pour la thérapie génique, bien que les décaissements restent sporadiques.
L'Europe maintient une part stable grâce au Réseau européen de référence pour les maladies métaboliques héréditaires qui coordonne les soins transfrontaliers. L'Allemagne valide la parité des coûts de la perfusion à domicile et soutient les renouvellements mensuels d'ordonnances pour minimiser les visites en clinique. La région Lombardie en Italie rend obligatoire le dépistage néonatal des maladies lysosomales de surcharge, élargissant l'adoption du diagnostic précoce. La France privilégie les modèles de remboursement liés aux résultats pour les thérapies géniques à coût élevé, pilotant des clauses de garantie de cinq ans. Le Service national de santé du Royaume-Uni (NHS) négocie des paiements par abonnement inspirés de son contrat antimicrobien de type « Netflix », visant à plafonner les dépenses annuelles en thérapies pour les maladies lysosomales de surcharge tout en garantissant le volume au fournisseur.

Paysage concurrentiel
L'unité Genzyme de Sanofi ancre le leadership dans la thérapie enzymatique substitutive avec les franchises Cerezyme, Fabrazyme et Myozyme dépassant 1,8 milliard USD de ventes combinées en 2024. Takeda maintient sa profondeur dans les mucopolysaccharidoses, tandis que VIMIZIM et NAGLAZYME de BioMarin ont généré 2,4 milliards USD de revenus en 2023, finançant l'expansion du pipeline dans la thérapie génique. Les acteurs historiques du marché reformulent les enzymes de première génération avec des technologies de PEGylation et de glyco-ingénierie pour contrer la pression des biosimilaires.
Orchard Therapeutics a acquis l'avantage du premier entrant dans la thérapie génique ciblant le système nerveux central avec Lenmeldy, avec un prix de 4,25 millions USD dans le cadre du déploiement d'accords basés sur la valeur. UX111 d'Ultragenyx est en tête du calendrier réglementaire 2025 avec une révision prioritaire de la FDA pour le syndrome de Sanfilippo. REGENXBIO co-développe RGX-121 au Japon dans le cadre d'un accord de 110 millions USD avec Nippon Shinyaku, illustrant la mondialisation des stratégies de commercialisation.
Les plateformes de traversée de la barrière hémato-encéphalique sont un foyer d'activité partenariale ; Chiesi a concédé la licence de la navette à médiation par anticorps d'Aliada pour la fusionner avec des enzymes lysosomales pour les indications neuronopathiques. La capacité de fabrication de vecteurs viraux émerge comme un différenciateur stratégique : les entreprises disposant de suites de bioréacteurs de 2 000 L en interne rapportent un coût des marchandises vendues inférieur de 35 % à celui des pairs dépendant des organisations de développement et de fabrication sous contrat (CDMO). Les indications en espace vierge telles que la gangliosidose GM1 attirent des investissements en phase précoce ; la thérapie génique préclinique de GC Biopharma a réduit les niveaux de GM1 de plus de 70 %, signalant la profondeur du pipeline au-delà des trois maladies « principales » traditionnelles.
Leaders du secteur du traitement des maladies lysosomales de surcharge
Pfizer Inc
Takeda Pharmaceutical Company Limited (Shire Plc)
Sanofi (Genzyme Corporation)
BioMarin
Johnson & Johnson (Actelion Pharmaceuticals Ltd)
- *Avis de non-responsabilité : les principaux acteurs sont triés sans ordre particulier

Développements récents dans le secteur
- Juillet 2025 : BioMarin a finalisé son acquisition d'Inozyme pour 270 millions USD, ajoutant la thérapie enzymatique en phase avancée INZ-701 pour la déficience en ENPP1 à son portefeuille de maladies rares.
- Février 2025 : Ultragenyx a reçu une révision prioritaire de la FDA pour la thérapie génique UX111 ciblant le syndrome de Sanfilippo de type A, avec une date PDUFA fixée au 18 août 2025.
- Janvier 2025 : REGENXBIO et Nippon Shinyaku ont annoncé un partenariat de 110 millions USD pour le co-développement de RGX-121 et RGX-111 pour les troubles MPS au Japon.
- Novembre 2024 : La FDA a approuvé Kebilidi, la première thérapie génique pour la déficience en AADC, administrée via le système de canule SmartFlow Neuro.
- Septembre 2024 : La FDA a autorisé Miplyffa et AQNEURSA comme premiers traitements pour la maladie de Niemann-Pick de type C, inaugurant des options de thérapie à double modalité.
- Mai 2024 : La FDA a autorisé Lenmeldy pour la leucodystrophie métachromatique, le tariffant à 4,25 millions USD et attribuant un bon de révision prioritaire à Orchard Therapeutics.


Portée du rapport mondial sur le marché du traitement des maladies lysosomales de surcharge
Selon le périmètre du rapport, les maladies lysosomales de surcharge (MLS) sont des erreurs innées du métabolisme caractérisées par l'accumulation de substrats en excès dans les cellules de divers organes due au fonctionnement défectueux des lysosomes. Elles provoquent un dysfonctionnement des organes où elles s'accumulent et contribuent à une morbidité et une mortalité élevées. Le marché du traitement des maladies lysosomales de surcharge est segmenté par type de thérapie (thérapie enzymatique substitutive, thérapie de réduction du substrat), par application (maladie de Gaucher, cystinose, maladie de Pompe, maladie de Fabry et autres applications) et par géographie (Amérique du Nord, Europe, Asie-Pacifique et reste du monde). Le rapport offre la valeur (en millions USD) pour les segments ci-dessus.
| Thérapie enzymatique substitutive (ERT) |
| Thérapie de réduction du substrat (SRT) |
| Thérapie génique |
| Thérapie par chaperon pharmacologique |
| Transplantation de cellules souches hématopoïétiques |
| Biothérapies intraveineuses |
| Petites molécules orales |
| Administration intrathécale/intracérébroventriculaire |
| Maladie de Gaucher (types I à III) |
| Maladie de Fabry |
| Maladie de Pompe |
| Mucopolysaccharidoses (I, II, III, IV, VI, VII) |
| Maladie de Niemann-Pick (types A/B et C) |
| Autres types de maladies |
| Perfusion IV en milieu hospitalier |
| Perfusion IV à domicile |
| Administration orale |
| Administration intrathécale/intracérébroventriculaire |
| Hôpitaux tertiaires |
| Cliniques spécialisées/maladies rares |
| Environnements de soins à domicile |
| Amérique du Nord | États-Unis |
| Canada | |
| Mexique | |
| Europe | Allemagne |
| Royaume-Uni | |
| France | |
| Italie | |
| Espagne | |
| Reste de l'Europe | |
| Asie-Pacifique | Chine |
| Japon | |
| Inde | |
| Australie | |
| Corée du Sud | |
| Reste de l'Asie-Pacifique | |
| Moyen-Orient et Afrique | Conseil de coopération du Golfe (CCG) |
| Afrique du Sud | |
| Reste du Moyen-Orient et de l'Afrique | |
| Amérique du Sud | Brésil |
| Argentine | |
| Reste de l'Amérique du Sud |
| Par type de thérapie | Thérapie enzymatique substitutive (ERT) | |
| Thérapie de réduction du substrat (SRT) | ||
| Thérapie génique | ||
| Thérapie par chaperon pharmacologique | ||
| Transplantation de cellules souches hématopoïétiques | ||
| Par modalité | Biothérapies intraveineuses | |
| Petites molécules orales | ||
| Administration intrathécale/intracérébroventriculaire | ||
| Par type de maladie | Maladie de Gaucher (types I à III) | |
| Maladie de Fabry | ||
| Maladie de Pompe | ||
| Mucopolysaccharidoses (I, II, III, IV, VI, VII) | ||
| Maladie de Niemann-Pick (types A/B et C) | ||
| Autres types de maladies | ||
| Par voie d'administration | Perfusion IV en milieu hospitalier | |
| Perfusion IV à domicile | ||
| Administration orale | ||
| Administration intrathécale/intracérébroventriculaire | ||
| Par utilisateur final | Hôpitaux tertiaires | |
| Cliniques spécialisées/maladies rares | ||
| Environnements de soins à domicile | ||
| Géographie | Amérique du Nord | États-Unis |
| Canada | ||
| Mexique | ||
| Europe | Allemagne | |
| Royaume-Uni | ||
| France | ||
| Italie | ||
| Espagne | ||
| Reste de l'Europe | ||
| Asie-Pacifique | Chine | |
| Japon | ||
| Inde | ||
| Australie | ||
| Corée du Sud | ||
| Reste de l'Asie-Pacifique | ||
| Moyen-Orient et Afrique | Conseil de coopération du Golfe (CCG) | |
| Afrique du Sud | ||
| Reste du Moyen-Orient et de l'Afrique | ||
| Amérique du Sud | Brésil | |
| Argentine | ||
| Reste de l'Amérique du Sud | ||


Questions clés traitées dans le rapport
Quelle est la taille actuelle du marché du traitement des maladies lysosomales de surcharge ?
La taille du marché du traitement des maladies lysosomales de surcharge a atteint 5,12 milliards USD en 2026 et devrait croître pour atteindre 7,39 milliards USD d'ici 2031.
Quel type de thérapie détient la plus grande part de marché ?
La thérapie enzymatique substitutive a maintenu 65,10 % des revenus mondiaux en 2025, ce qui en fait l'approche thérapeutique dominante.
Pourquoi la thérapie génique est-elle considérée comme perturbatrice dans ce domaine ?
La thérapie génique offre un bénéfice potentiellement curatif à dose unique et croît à un TCAC de 10,22 %, dépassant toutes les autres modalités.
Quelle région connaît la croissance la plus rapide ?
L'Asie-Pacifique devrait afficher un TCAC de 9,15 % jusqu'en 2031, portée par la modernisation réglementaire et l'expansion du dépistage néonatal.
Comment les coûts élevés des traitements sont-ils gérés ?
Les payeurs adoptent des contrats basés sur les résultats, des modèles d'abonnement et des paiements de type annuité pour répartir l'impact financier des thérapies dont le prix dépasse 4 millions USD.
Dernière mise à jour de la page le: