Marktgröße und Marktanteil für die Behandlung von lysosomalen Speicherkrankheiten
Marktübersicht
| Studienzeitraum | 2022 - 2031 |
|---|---|
| Marktgröße (2026) | 5.12 Milliarden US-Dollar |
| Marktgröße (2031) | 7.39 Milliarden US-Dollar |
| Wachstumsrate (2026 - 2031) | 7.65% CAGR |
| Schnellstwachsender Markt | Asien-Pazifik |
| Größter Markt | Nordamerika |
| Marktkonzentration | Mittel |
Hauptakteure *Haftungsausschluss: Hauptakteure in keiner bestimmten Reihenfolge sortiert Bild © Mordor Intelligence. Wiederverwendung erfordert Namensnennung gemäß CC BY 4.0. | |
Marktanalyse für die Behandlung von lysosomalen Speicherkrankheiten von Mordor Intelligence
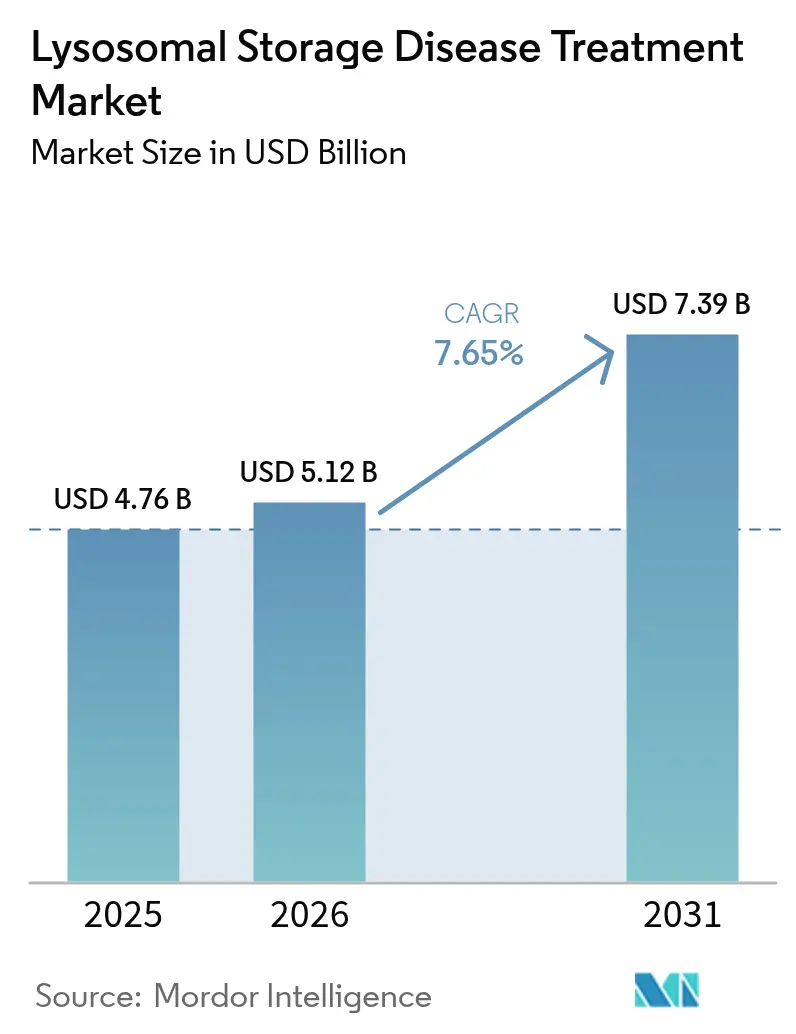
Die Marktgröße für die Behandlung von lysosomalen Speicherkrankheiten wurde im Jahr 2025 auf 4,76 Milliarden USD geschätzt und soll von 5,12 Milliarden USD im Jahr 2026 auf 7,39 Milliarden USD bis 2031 wachsen, mit einer CAGR von 7,65 % während des Prognosezeitraums (2026–2031). Die zunehmenden Zulassungen für Einmaldosis-Gentherapien, breitere Indikationen für die Enzymersatztherapie und genomische Neugeborenen-Screening-Programme haben den Behandlungsbeginn in präsymptomatische Stadien verlagert und die adressierbare Population erweitert. Der Wettbewerbsdruck steigt, da etablierte Enzymhersteller Herausforderern gegenüberstehen, die Blut-Hirn-Schranken-penetrierende Vektoren und orale kleinmolekulare Substrate vorantreiben. Die Hochskalierung der Herstellung viraler Vektoren bleibt ein Engpass, auch wenn Kapital in Auftragsentwicklungsorganisationen fließt. Das parallele Wachstum von Heiminfusionsdiensten, die die Verabreichungskosten um 25–50 % senken, gestaltet die Modelle der Versorgungserbringung neu.
Wichtigste Erkenntnisse des Berichts
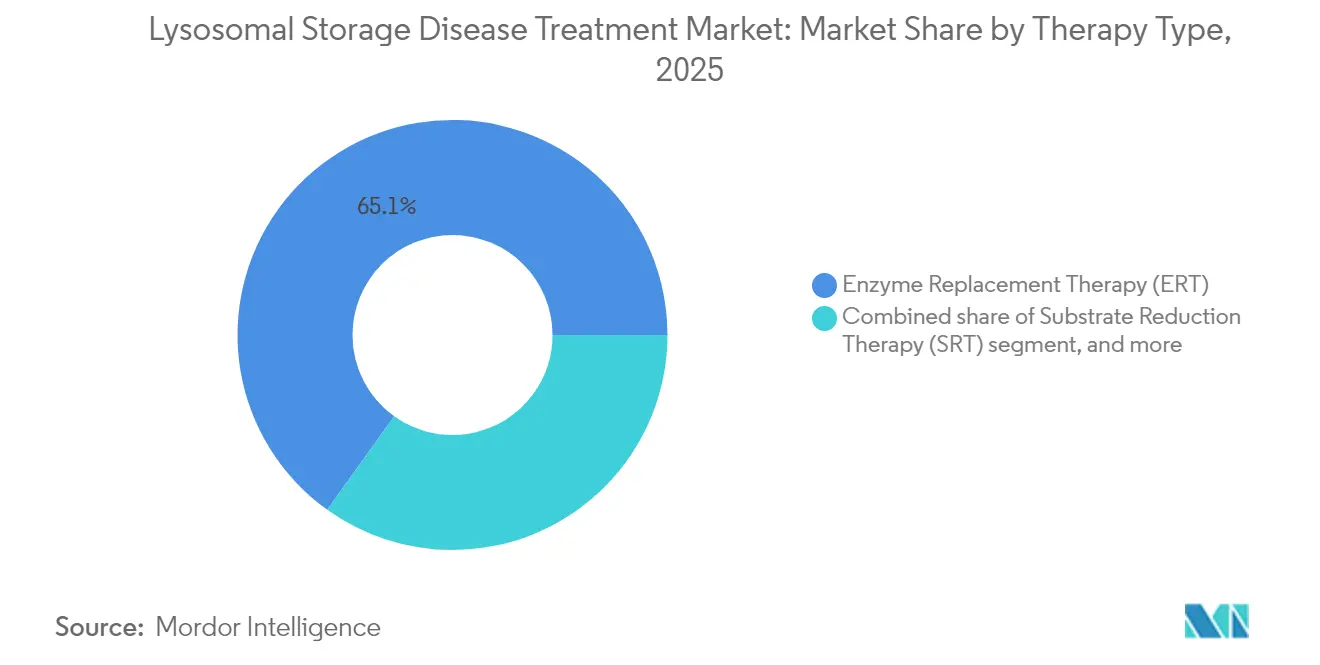
- Nach Therapietyp führte die Enzymersatztherapie mit einem Marktanteil von 65,10 % am Markt für die Behandlung von lysosomalen Speicherkrankheiten im Jahr 2025; die Gentherapie wird voraussichtlich bis 2031 mit einer CAGR von 10,22 % wachsen.
- Nach Modalität entfielen auf intravenöse Biologika 55,20 % des Marktanteils an der Marktgröße für die Behandlung von lysosomalen Speicherkrankheiten im Jahr 2025, während die orale kleinmolekulare Therapie bis 2031 voraussichtlich mit einer CAGR von 10,12 % wachsen wird.
- Nach Krankheitstyp entfiel auf die Gaucher-Krankheit ein Anteil von 28,10 % an der Marktgröße für die Behandlung von lysosomalen Speicherkrankheiten im Jahr 2025; die Niemann-Pick-Krankheit schreitet bis 2031 mit einer CAGR von 10,35 % voran.
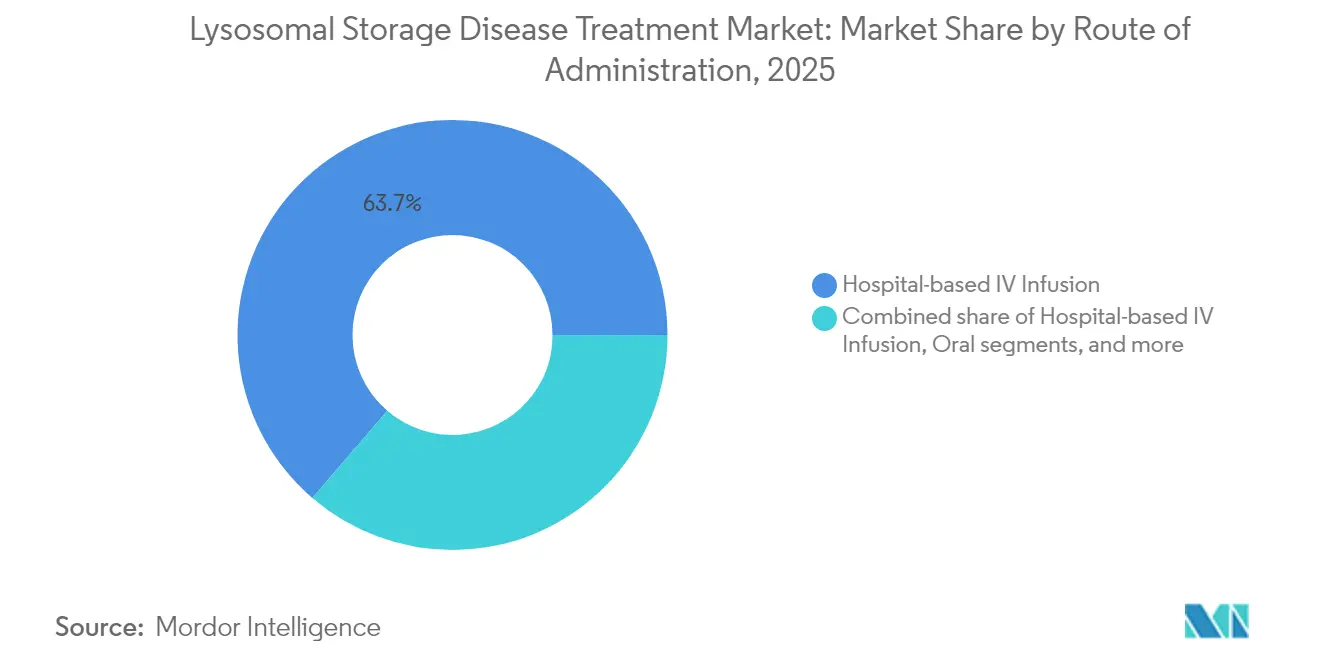
- Nach Verabreichungsweg hielt die krankenhausbasierte IV-Infusion 63,70 % des Marktanteils am Markt für die Behandlung von lysosomalen Speicherkrankheiten im Jahr 2025, während die Heim-IV-Infusion bis 2031 voraussichtlich eine CAGR von 11,05 % erzielen wird.
- Nach Endnutzer entfielen auf Tertiärkrankenhäuser 65,60 % des Umsatzanteils im Jahr 2025, während Heimversorgungseinrichtungen bis 2031 voraussichtlich mit einer CAGR von 11,20 % wachsen werden.
- Nach Geografie entfiel auf Nordamerika ein Umsatzanteil von 42,10 % im Jahr 2025, während der asiatisch-pazifische Raum bis 2031 voraussichtlich mit einer CAGR von 9,15 % wachsen wird.
Hinweis: Die Marktgrößen- und Prognosezahlen in diesem Bericht werden mithilfe des proprietären Schätzrahmens von Mordor Intelligence erstellt und mit den neuesten verfügbaren Daten und Erkenntnissen bis 2026 aktualisiert.
Globale Trends und Erkenntnisse zum Markt für die Behandlung von lysosomalen Speicherkrankheiten
Analyse der Treiberwirkungen*
| Treiber | Prozentualer Einfluss auf die CAGR-Prognose | Geografische Relevanz | Zeithorizont der Auswirkungen |
|---|---|---|---|
| Steigende weltweite Prävalenz von lysosomalen Speicherkrankheiten | +1.2% | Global – höchste Erkennung in entwickelten Märkten | Mittelfristig (2–4 Jahre) |
| Zunehmende Designierungen und Anreize für Orphan-Arzneimittel | +1.0% | Führungsrolle Nordamerika und EU; weltweite Ausweitung der Annahme | Kurzfristig (≤ 2 Jahre) |
| Fortschritte bei Diagnosetechnologien und Früherkennung | +1.8% | Nordamerika und EU führend; rasche Einführung in APAC | Kurzfristig (≤ 2 Jahre) |
| Ausweitung der Produktzulassungen für die Enzymersatztherapie | +1.3% | Global, am stärksten in Nordamerika und der EU | Mittelfristig (2–4 Jahre) |
| Aufkommende Gen- und Zelltherapien für neuropathische lysosomale Speicherkrankheiten | +2.1% | Global, konzentriert auf wichtige Pharmazeutika-Zentren | Langfristig (≥ 4 Jahre) |
| Strategische Kooperationen und Anstieg der Investitionen in seltene Krankheiten | +1.5% | Kern Nordamerika und EU; Ausstrahlungseffekt auf Schwellenmärkte | Mittelfristig (2–4 Jahre) |
| Quelle: Mordor Intelligence | |||
Steigende weltweite Prävalenz von lysosomalen Speicherkrankheiten
Das genomische Neugeborenenscreening in Shanghai identifizierte eine Inzidenz lysosomaler Speicherkrankheiten von 1 zu 1.856 Lebendgeburten, was wesentlich über den historischen Schätzungen liegt und den Wert proaktiver Tests bestätigt. Das CARE-Programm der chinesischen Nationalen Medizinprodukteadministration fördert Orphan-Drug-Anmeldungen und hat die Einreichungen von Investigational New Drug-Anträgen seit 2024 um 32 % gesteigert. Ähnliche Prävalenzanstiege sind im nationalen Register Südkoreas zu beobachten, was den Übergang von anekdotischer Diagnose zu bevölkerungsweitem Screening unterstreicht. Mit der Ausweitung der Register wächst die Nachfrage nach Frühstadium-Interventionen, was die Diagnoseverzögerung in führenden Tertiärzentren von sechs Jahren auf weniger als sechs Wochen verkürzt. Die therapeutischen Pipelines werden daher neu auf Indikationen mit Beginn im Säuglingsalter ausgerichtet, bei denen der klinische Nutzen am ausgeprägtesten ist.
Fortschritte bei Diagnosetechnologien und Früherkennung
Die Tandem-Massenspektrometrie der zweiten Generation scannt nun in einem einzigen Assay auf bis zu acht lysosomale Speicherkrankheiten mit unter 0,5 % Falsch-Positiv-Rate und übertrifft damit Enzymaktivitätspanels. Die Integration der Sequenzierung der nächsten Generation bestätigt pathogene Varianten und leitet Präzisionstherapien, wie die Auswahl von Migalastat für Fabry-Patienten mit geeigneten Mutationen. Die Bearbeitungszeiten für bestätigende Genotypisierungen sind auf unter sieben Tage gefallen, was die Behandlungseinleitung während neonatologischer Intensivaufenthalte ermöglicht. Die Akzeptanz im Gesundheitssystem ist in Kanada und Deutschland am schnellsten, wo die staatliche Erstattung sowohl biochemische als auch genomische Assays abdeckt. Die Frühdiagnose katalysiert die Akzeptanz hochpreisiger Gentherapien durch Kostenträger, indem sie langfristige Kostenausgleiche durch vermiedene Behinderungen belegt.
Aufkommende Gen- und Zelltherapien für neuropathische lysosomale Speicherkrankheiten
Lenmeldy's Einmaldosis-autologe Stammzelltherapie erzielte motorische Meilensteinerhaltung bei 90 % der behandelten Patienten mit metachromatischer Leukodystrophie gegenüber 30 %, die historisch in unbehandelten Kohorten beobachtet wurden[1]US-amerikanische Behörde für Lebens- und Arzneimittel (FDA), "Programm für vorrangige Überprüfungsgutscheine für seltene pädiatrische Krankheiten," fda.gov. RGX-121 senkte das Heparansulfat im Liquor cerebrospinalis um 85 % und ermöglichte es 80 % der Patienten, wöchentliche Infusionen einzustellen. AAV9-Kapsiden penetrieren die Blut-Hirn-Schranke effizienter als frühere Vektoren und erhalten die Enzymexpression in Gaucher- und GM1-Gangliosidose-Studien über mindestens 36 Monate aufrecht. Herstellungsausbeuten, einst auf 1e15 Vektorgenom pro Charge begrenzt, haben sich durch Suspensions-HEK293-Technologien verdoppelt, was die Herstellungskosten um 38 % senkt. Regulierungsbehörden führen Rolling-Review-Mechanismen ein, die die Zulassungsfristen durchschnittlich um neun Monate verkürzt haben, was den Wettbewerbsdruck intensiviert.
Strategische Kooperationen und Anstieg der Investitionen in seltene Krankheiten
REGENXBIOs Lizenzierungsdeal über 110 Millionen USD mit Nippon Shinyaku für MPS-Programme ist ein Beispiel für eine Verlagerung hin zu regionalen Co-Vermarktungsstrukturen, die das Risiko streuen und den Markteintritt beschleunigen. BioMarins Erwerb von Inozyme für 270 Millionen USD sichert eine spätphasige Enzymersatztherapie für ENPP1-Mangel und festigt die therapeutische Portfoliobreite. Die Risikokapitalfinanzierung bleibt widerstandsfähig, wobei Glycomine 115 Millionen USD in der Serie C sichert, um Therapeutika für Glykosylierungsstörungen voranzutreiben. Partnerschaften konzentrieren sich nun auf Plattformen für Blut-Hirn-Schranken-Shuttles und modulares Kapsidengineering, Bereiche, die bis 2027 voraussichtlich mehr als 2 Milliarden USD an neuem Kapital anziehen werden. Solche Allianzen verkürzen die F&E-Zeiträume und gewähren kleineren Biotechnologieunternehmen Zugang zu etablierter Fertigungs- und Regulierungskompetenz.
Analyse der Hemmfaktorwirkungen*
| Analyse der Hemmfaktorwirkungen | (~) Prozentualer Einfluss auf die CAGR-Prognose | Geografische Relevanz | Zeithorizont der Auswirkungen |
|---|---|---|---|
| Hohe Behandlungskosten und Erstattungsherausforderungen | −1.8% | Global – am akutesten in Schwellenmärkten | Langfristig (≥ 4 Jahre) |
| Begrenzte Penetration der Blut-Hirn-Schranke durch aktuelle Therapien | −1.2% | Global – betrifft neuronopathische Formen | Mittelfristig (2–4 Jahre) |
| Skalierbarkeit der Herstellung und Versorgungsengpässe | −1.4% | Global, konzentriert auf Produktionszentren für Gentherapien | Mittelfristig (2–4 Jahre) |
| Langzeitbedenken hinsichtlich Sicherheit und Immunogenität | −1.0% | Global, betrifft alle fortgeschrittenen Modalitäten | Langfristig (≥ 4 Jahre) |
| Quelle: Mordor Intelligence | |||
Hohe Behandlungskosten und Erstattungsherausforderungen
Lenmeldy wird zu einem Preis von 4,25 Millionen USD angeboten, was es zur teuersten Therapie der Welt macht und die Kostenträger zwingt, ergebnisbasierte Verträge zu verabschieden, die die Erstattung an motorische Endpunkte knüpfen[2]Thomas Mueller et al., "Kostenbelastung der Enzymersatztherapie in Deutschland," Orphanet Journal of Rare Diseases, ojrd.biomedcentral.com. In Deutschland betragen die durchschnittlichen jährlichen Enzymersatztherapieausgaben für die Fabry-Krankheit 369.047 EUR (405.952 USD) pro Patient, wovon 94 % auf Arzneimittelerwerbskosten entfallen. Medicaid-Programme gliedern Gentherapie-Pakete aus Kopfpauschalen-Zahlungen aus, um einmalige finanzielle Schocks zu mildern. Private Versicherungsträger schränken die Kostenübernahme auf genetisch bestätigte Fälle ein und fordern longitudinale Biomarkerdaten, was den Zugang in 17 US-amerikanischen Bundesstaaten verlängert. Innovative Rentenmodell-Zahlungsmodelle schreiten aufgrund ungeklärter regulatorischer Leitlinien zur mehrjährigen Amortisation langsam voran.
Begrenzte Penetration der Blut-Hirn-Schranke durch aktuelle Therapien
Intravenöses Imiglucerase überwindet die Blut-Hirn-Schranke nicht, wodurch die neuronopathische Gaucher-Krankheit trotz Kontrolle somatischer Symptome unbehandelt bleibt. Rezeptor-vermittelte Transzytose mit Transferrin-Liganden zeigte eine vierfache Enzymaufnahme in präklinischen Pompe-Modellen, wartet jedoch noch auf Phase-2-Bestätigung. Intrathekales Cerliponase alfa verlängert das Überleben bei CLN2-Patienten, erfordert jedoch vierteljährlichen neurochirurgischen Katheterzugang, was die breite Anwendung einschränkt. Genvektoren zeigen überlegenen ZNS-Tropismus, wobei neutralisierende Antikörper gegen AAV9 bis zu 25 % der Erwachsenen vom Studieneinschluss ausschließen. Herstellungsengpässe für hochtitrierte Vektoren bestehen trotz Dual-Plasmid-Produzentenlinien, die die Ausbeute um 30 % steigern, weiter fort.
*Unsere Prognosen behandeln die Auswirkungen von Treibern und Einschränkungen als richtungsweisend und nicht additiv. Die Wirkungsprognosen berücksichtigen Basiswachstum, Mischungseffekte und Wechselwirkungen zwischen Variablen.
Segmentanalyse
Nach Therapietyp: Anhaltende Führungsposition der Enzymersatztherapie trifft auf rasanten Aufstieg der Gentherapie
Die Marktgröße für die Behandlung von lysosomalen Speicherkrankheiten für die Enzymersatztherapie betrug im Jahr 2025 3,1 Milliarden USD, entsprechend 65,10 % des Gesamtumsatzes, und bleibt maßgeblich für die Behandlung von Gaucher-, Fabry- und Pompe-Erkrankungen. Die Wettbewerbspositionierung konzentriert sich auf Enzyme der zweiten Generation wie Avalglucosidase alfa, das eine höhere Muskelaufnahme als das Vorgängerprodukt Alglucosidase bietet. Die CAGR von 10,22 % der Gentherapie spiegelt jedoch ihr Wertversprechen der Einmaldosierung wider. Lenmeldy's Zulassung demonstriert dauerhaften neurologischen Nutzen, während Ultragenyx's UX111 die FDA-Prüfwarteschlange 2025 für das Sanfilippo-Syndrom anführt. Substratreduktion und pharmakologische Chaperone liefern inkrementelle Gewinne für mutationsspezifische Kohorten und stärken Präzisionsmedizin-Trajektorien innerhalb des Marktes für die Behandlung von lysosomalen Speicherkrankheiten. Die Pipeline-Konvergenz hin zu Kombinationsregimen, wie Pombiliti plus Opfolda, zielt darauf ab, den intrazellulären Transport zu verstärken und Behandlungsintervalle zu verlängern.
Notiz: Segmentanteile aller einzelnen Segmente sind nach Berichtkauf verfügbar


Nach Modalität: Orale Regime gewinnen gegenüber parenteralen Standards an Boden
Intravenöse Biologika erzielten 2025 einen Umsatzanteil von 55,20 %, getragen durch wöchentliche Infusionen von Imiglucerase, Agalsidase beta und Alglucosidase alfa. Orale kleinmolekulare Substratreduktoren – Venglustat, Lucerastat und das zugelassene Cerdelga – verzeichnen mit einer CAGR von 10,12 % das stärkste Wachstum, getrieben durch Adhärenzvorteile und das Fehlen von Infusionsreaktionen. Der Modalitätsmix verschiebt sich weiter, da der orale Chaperon Migalastat auf zusätzliche Fabry-Genotypen ausgeweitet wird. Intrathekale und intrazerebroventrikuäre Verabreichungen sind neuropathischen Syndromen vorbehalten; Geräte wie SmartFlow Neuro ermöglichen eine präzise Kanülenplatzierung für den Kebilidi-Gentransfer. PEGylierung und Fc-Fusionstechnologien verlängern die Halbwertszeit von Enzymersatztherapien wie ELFABRIO und reduzieren die Infusionshäufigkeit auf monatlich. Die patientenzentrierte Dosierung steht im Einklang mit der Ausweitung der Heiminfusion und unterstreicht den Fokus der Kostenträger auf die Reduzierung der Gesamtbehandlungskosten innerhalb des Marktes für die Behandlung von lysosomalen Speicherkrankheiten.
Nach Krankheitstyp: Etabliertes Gaucher-Portfolio gegenüber raschem Niemann-Pick-Momentum
Die Gaucher-Krankheit bleibt der größte Teilmarkt mit 28,10 % des Umsatzes im Jahr 2025, unterstützt durch drei kommerzielle Enzymersatztherapien und ein orales Präparat, die den Preisdruck diversifizieren. Der Wettbewerb treibt periodische Preisnachlässe und erweiterte Programme zur compassionate use an. Die Niemann-Pick-Krankheit steht exemplarisch für die zukünftige Hochwachstumskohorte mit einer CAGR von 10,35 % bis 2031, nachdem die doppelte FDA-Zulassung von Miplyffa (oral) und AQNEURSA (IV) Jahrzehnte therapeutischer Leere beendete. Fabry und Pompe verzeichnen ein mittleres einstelliges Wachstum dank Chaperon-Therapie-Übernahme und Enzymen der nächsten Generation mit überlegenen renalen und pulmonalen Endpunkten. Mukopolysaccharidosen treiben Gentherapieinnovationen voran; RGX-111 für MPS IIB könnte dauerhafte ZNS-Korrektur liefern und die chronische intrathekale Enzymersatztherapie herausfordern. Aufkommende Pipelines bei GM1-Gangliosidose und Alpha-Mannosidose veranschaulichen das Potenzial für unbesetzte Marktnischen, da frühphasige Daten eine Reduktion von Speichersubstraten um mehr als 70 % zeigen.
Nach Verabreichungsweg: Heiminfusion zeichnet neue Versorgungspfade
Krankenhausbasierte IV-Zentren lieferten 2025 63,70 % der Dosen, doch die Steuerung der Kostenträger hin zu kostengünstigeren Versorgungsorten beschleunigt die Verlagerung in häusliche Einrichtungen, die nun eine CAGR von 11,05 % verzeichnen. Reale Belege aus deutschen Fabry-Kohorten zeigen eine Parität bei den Nebenwirkungsraten zwischen Heim- und Klinikinfusionen, während die nichtmedikamentösen Kosten um 42 % gesenkt werden. Orale Regime treiben die Dezentralisierung weiter voran und ermöglichen die Selbstverabreichung und die Verringerung von Reisebelastungen für ländliche Familien. Intrathekale Wege werden trotz chirurgischer Anforderungen zunehmend in Satelliten-Neurochirurgiezentren angeboten, um den Zugang zu erweitern. Gentherapien, die einmalig in spezialisierten Krankenhäusern verabreicht werden, können anschließend den Bedarf an chronischen Infusionen obsolet machen und unterstreichen die disruptiven Auswirkungen auf die Versorgungsinfrastruktur innerhalb des Marktes für die Behandlung von lysosomalen Speicherkrankheiten.
Notiz: Segmentanteile aller einzelnen Segmente sind nach Berichtkauf verfügbar
Nach Endnutzer: Tertiärzentren verlieren die Exklusivität zugunsten gemeinschaftsbasierter Versorgung
Tertiärkrankenhäuser betreuen aufgrund komplexer Diagnose- und Überwachungsanforderungen nach wie vor 65,60 % der behandelten Patienten. Dennoch verzeichnen Heimversorgungsprogramme, unterstützt durch Telemedizin, eine CAGR von 11,20 %, da pflegegeleitete Infusionsteams ihre geografische Reichweite ausweiten. Spezialkliniken für seltene Krankheiten, oft universitär angegliedert, bewahren ihre Relevanz durch den Zugang zu experimentellen Protokollen und multidisziplinärer Expertise. Die Einmaldosierung in der Gentherapie komprimiert die langfristigen Krankenhausumsatzströme und veranlasst Einrichtungen, wertbasierte Verträge und ergänzende genomische Beratungsleistungen zu suchen. Kostenträger erproben integrierte Versorgungspakete, die genetische Tests, Beratung und Arzneimittellieferung unter einem einzigen Code erstatten und so die Anreize auf das Gemeinschaftsmanagement im Markt für die Behandlung von lysosomalen Speicherkrankheiten ausrichten.
Geografische Analyse
Nordamerika lieferte 2025 42,10 % des Umsatzes, geleitet durch das Programm für vorrangige Überprüfungsgutscheine für seltene pädiatrische Krankheiten der US-amerikanischen Behörde für Lebens- und Arzneimittel (FDA) und eine weitreichende kommerzielle Krankenversicherungsdeckung. Mehr als 20 aktive Gentherapiestudien rekrutieren in den Vereinigten Staaten und festigen die Innovationsführerschaft der Region. Disparitäten auf Staatsebene bestehen weiter: Kalifornien übernimmt Reise- und Unterkunftskosten für Lenmeldy-Empfänger aus anderen Bundesstaaten, während sechs Midwestern-Staaten Gentherapien auf Medicaid-Auszahlungen beschränken und so die Zugangsfristen verlängern. Kanadas universelles System erstattet fünf Enzymersatztherapien auf einer eigens ausgewiesenen Orphan-Drug-Liste, wobei die Verzögerung zwischen Zulassung und Finanzierung im Durchschnitt 14 Monate beträgt.
Der asiatisch-pazifische Raum ist mit einer CAGR von 9,15 % das am schnellsten wachsende Gebiet. Chinas aktualisierte Liste seltener Krankheiten, die nun 207 Erkrankungen umfasst, hat die behördliche Prüfungszeit für importierte Präparate auf 9 Monate verkürzt, gegenüber 24 Monaten vor 2023. Das genomische Screening-Pilotprojekt in Shanghai hat eine LSD-Inzidenz weit über dem globalen Durchschnitt aufgedeckt und nationale Skalierungszuschüsse ausgelöst. Japan nutzt seinen Sakigake-Fast-Track, um ausländische Biotechnologieanmeldungen anzuziehen, während Südkoreas Nationale Krankenversicherung über 90 % der Fabry-Behandlungskosten abdeckt und damit eine fortgeschrittene Erstattungsbereitschaft demonstriert. Indiens Politik für seltene Krankheiten subventioniert bis zu 2 Millionen INR (2,2 Millionen USD) pro Patient für Gentherapien, wobei die Auszahlung jedoch sporadisch bleibt.
Europa hält einen stabilen Anteil, getragen durch das Europäische Referenznetzwerk für hereditäre Stoffwechselstörungen, das grenzüberschreitende Versorgung koordiniert. Deutschland validiert die Kostenparität der Heiminfusion und unterstützt monatliche Rezeptnachfüllungen, um Klinikbesuche zu minimieren. Die Region Lombardei in Italien schreibt das Neugeborenenscreening auf lysosomale Speicherkrankheiten vor und erweitert die Frühdiagnose. Frankreich bevorzugt ergebnisgebundene Erstattungsmodelle für hochpreisige Gentherapien und erprobt Fünfjahres-Garantieklauseln. Der britische Nationale Gesundheitsdienst (NHS) verhandelt Abonnementzahlungen, die nach seinem antimikrobiellen „Netflix”-Vertrag modelliert sind, um die jährlichen Ausgaben für lysosomale Speicherkrankheitstherapien zu begrenzen und gleichzeitig das Lieferantenvolumen zu garantieren.

Wettbewerbslandschaft
Sanofis Genzyme-Einheit verankert die Führungsposition in der Enzymersatztherapie mit den Franchises Cerezyme, Fabrazyme und Myozyme, deren kombinierter Umsatz im Jahr 2024 1,8 Milliarden USD übersteigt. Takeda hält ein breites Portfolio bei Mukopolysaccharidosen, während BioMarins VIMIZIM und NAGLAZYME 2023 einen Umsatz von 2,4 Milliarden USD erzielten und die Pipeline-Erweiterung in die Gentherapie finanzieren. Marktführer reformulieren Enzyme der ersten Welle mit PEGylierung und Glykotechnik, um dem Biosimilar-Druck entgegenzuwirken.
Orchard Therapeutics erlangte den First-Mover-Vorteil bei ZNS-zielgerichteter Gentherapie mit Lenmeldy und einem Preis von 4,25 Millionen USD inmitten der Einführung wertbasierter Vereinbarungen. Ultragenyx's UX111 führt den regulatorischen Kalender 2025 mit einer FDA-Prioritätsprüfung für das Sanfilippo-Syndrom an. REGENXBIO entwickelt RGX-121 gemeinsam in Japan im Rahmen eines 110-Millionen-USD-Pakts mit Nippon Shinyaku und veranschaulicht die Globalisierung von Vermarktungsstrategien.
Plattformen zur Überwindung der Blut-Hirn-Schranke sind ein Brennpunkt von Partnerschaftsaktivitäten; Chiesi lizenzierte Aliadas Antikörper-vermittelten Shuttle zur Fusion mit lysosomalen Enzymen für neuropathische Indikationen. Die Kapazität zur Herstellung viraler Vektoren entwickelt sich zu einem strategischen Differenzierungsmerkmal: Unternehmen mit eigenen 2.000-L-Bioreaktor-Suiten berichten von 35 % niedrigeren Herstellungskosten als Wettbewerber, die auf Auftragsfertigungsorganisationen angewiesen sind. Unbesetzte Indikationen wie GM1-Gangliosidose ziehen Frühphasen-Investitionen an; GC Biopharma's präklinische Gentherapie reduzierte GM1-Spiegel um mehr als 70 % und signalisiert die Pipeline-Breite jenseits der traditionellen „großen drei” Erkrankungen.
Marktführer in der Branche für die Behandlung von lysosomalen Speicherkrankheiten
Pfizer Inc
Takeda Pharmaceutical Company Limited (Shire Plc)
Sanofi (Genzyme Corporation)
BioMarin
Johnson & Johnson (Actelion Pharmaceuticals Ltd)
- *Haftungsausschluss: Hauptakteure in keiner bestimmten Reihenfolge sortiert

Jüngste Branchenentwicklungen
- Juli 2025: BioMarin schloss die Übernahme von Inozyme für 270 Millionen USD ab und fügte die spätphasige Enzymtherapie INZ-701 für ENPP1-Mangel seinem Portfolio für seltene Krankheiten hinzu.
- Februar 2025: Ultragenyx erhielt eine FDA-Prioritätsprüfung für die UX111-Gentherapie gegen das Sanfilippo-Syndrom Typ A, mit dem PDUFA-Datum auf den 18. August 2025 festgelegt.
- Januar 2025: REGENXBIO und Nippon Shinyaku gaben eine Partnerschaft über 110 Millionen USD bekannt, um RGX-121 und RGX-111 für MPS-Erkrankungen in Japan gemeinsam zu entwickeln.
- November 2024: Die US-amerikanische Behörde für Lebens- und Arzneimittel (FDA) genehmigte Kebilidi als erste Gentherapie für den AADC-Mangel, verabreicht über das SmartFlow Neuro Kanülensystem.
- September 2024: Die FDA genehmigte Miplyffa und AQNEURSA als erste Behandlungen für die Niemann-Pick-Krankheit Typ C und eröffnete damit duale Therapieoptionen.
- Mai 2024: Die FDA genehmigte Lenmeldy für die metachromatische Leukodystrophie zu einem Preis von 4,25 Millionen USD und vergab einen Priority Review Voucher an Orchard Therapeutics.


Umfang des globalen Marktberichts für die Behandlung von lysosomalen Speicherkrankheiten
Gemäß dem Berichtsumfang sind lysosomale Speicherkrankheiten (LSDs) angeborene Stoffwechselstörungen, die durch die übermäßige Ansammlung von Substraten in Zellen verschiedener Organe aufgrund der defekten Funktion von Lysosomen gekennzeichnet sind. Sie verursachen eine Dysfunktion jener Organe, in denen sie sich ansammeln, und tragen erheblich zu Morbidität und Mortalität bei. Der Markt für die Behandlung von lysosomalen Speicherkrankheiten ist segmentiert nach Therapietyp (Enzymersatztherapie, Substratreduktionstherapie), nach Anwendungsgebiet (Gaucher-Krankheit, Zystinose, Pompe-Krankheit, Fabry-Krankheit und weitere Anwendungsgebiete) sowie nach Geografie (Nordamerika, Europa, asiatisch-pazifischer Raum und Rest der Welt). Der Bericht bietet den Wert (in Millionen USD) für die oben genannten Segmente.
| Enzymersatztherapie (ERT) |
| Substratreduktionstherapie (SRT) |
| Gentherapie |
| Pharmakologische Chaperon-Therapie |
| Hämatopoetische Stammzelltransplantation |
| Intravenöse Biologika |
| Orales Kleinmolekül |
| Intrathekale/IZV-Verabreichung |
| Gaucher-Krankheit (Typ I–III) |
| Fabry-Krankheit |
| Pompe-Krankheit |
| Mukopolysaccharidosen (I, II, III, IV, VI, VII) |
| Niemann-Pick-Krankheit (Typ A/B & C) |
| Andere Krankheitstypen |
| Krankenhausbasierte IV-Infusion |
| Heim-IV-Infusion |
| Oral |
| Intrathekal/IZV |
| Tertiärkrankenhäuser |
| Spezialkliniken für seltene Krankheiten |
| Heimversorgungseinrichtungen |
| Nordamerika | Vereinigte Staaten |
| Kanada | |
| Mexiko | |
| Europa | Deutschland |
| Vereinigtes Königreich | |
| Frankreich | |
| Italien | |
| Spanien | |
| Rest Europas | |
| Asiatisch-pazifischer Raum | China |
| Japan | |
| Indien | |
| Australien | |
| Südkorea | |
| Rest des asiatisch-pazifischen Raums | |
| Naher Osten und Afrika | Golfkooperationsrat (GCC) |
| Südafrika | |
| Rest des Nahen Ostens und Afrikas | |
| Südamerika | Brasilien |
| Argentinien | |
| Rest Südamerikas |
| Nach Therapietyp | Enzymersatztherapie (ERT) | |
| Substratreduktionstherapie (SRT) | ||
| Gentherapie | ||
| Pharmakologische Chaperon-Therapie | ||
| Hämatopoetische Stammzelltransplantation | ||
| Nach Modalität | Intravenöse Biologika | |
| Orales Kleinmolekül | ||
| Intrathekale/IZV-Verabreichung | ||
| Nach Krankheitstyp | Gaucher-Krankheit (Typ I–III) | |
| Fabry-Krankheit | ||
| Pompe-Krankheit | ||
| Mukopolysaccharidosen (I, II, III, IV, VI, VII) | ||
| Niemann-Pick-Krankheit (Typ A/B & C) | ||
| Andere Krankheitstypen | ||
| Nach Verabreichungsweg | Krankenhausbasierte IV-Infusion | |
| Heim-IV-Infusion | ||
| Oral | ||
| Intrathekal/IZV | ||
| Nach Endnutzer | Tertiärkrankenhäuser | |
| Spezialkliniken für seltene Krankheiten | ||
| Heimversorgungseinrichtungen | ||
| Geografie | Nordamerika | Vereinigte Staaten |
| Kanada | ||
| Mexiko | ||
| Europa | Deutschland | |
| Vereinigtes Königreich | ||
| Frankreich | ||
| Italien | ||
| Spanien | ||
| Rest Europas | ||
| Asiatisch-pazifischer Raum | China | |
| Japan | ||
| Indien | ||
| Australien | ||
| Südkorea | ||
| Rest des asiatisch-pazifischen Raums | ||
| Naher Osten und Afrika | Golfkooperationsrat (GCC) | |
| Südafrika | ||
| Rest des Nahen Ostens und Afrikas | ||
| Südamerika | Brasilien | |
| Argentinien | ||
| Rest Südamerikas | ||


Im Bericht beantwortete Schlüsselfragen
Wie groß ist der aktuelle Markt für die Behandlung von lysosomalen Speicherkrankheiten?
Der Markt für die Behandlung von lysosomalen Speicherkrankheiten erreichte im Jahr 2026 einen Wert von 5,12 Milliarden USD und soll bis 2031 auf 7,39 Milliarden USD wachsen.
Welcher Therapietyp hält den größten Marktanteil?
Die Enzymersatztherapie hielt 2025 65,10 % des globalen Umsatzes und ist damit der dominante Behandlungsansatz.
Warum gilt die Gentherapie in diesem Bereich als disruptiv?
Die Gentherapie bietet einen Einmaldosis-Nutzen mit potenziell heilender Wirkung und wächst mit einer CAGR von 10,22 %, was alle anderen Modalitäten übertrifft.
Welche Region wächst am schnellsten?
Der asiatisch-pazifische Raum wird bis 2031 voraussichtlich eine CAGR von 9,15 % verzeichnen, getrieben durch regulatorische Modernisierung und die Ausweitung des Neugeborenenscreenings.
Wie werden die hohen Behandlungskosten gehandhabt?
Kostenträger verabschieden ergebnisbasierte Verträge, Abonnementmodelle und Rentenmodell-Zahlungen, um die finanzielle Auswirkung von Therapien mit einem Preis von über 4 Millionen USD zu verteilen.
Seite zuletzt aktualisiert am: