Taille et part du marché des CRO de dispositifs médicaux
VUE D’ENSEMBLE DU MARCHÉ
| Période d'étude | 2020 - 2031 |
|---|---|
| Taille du Marché (2026) | 11.91 Milliards de dollars |
| Taille du Marché (2031) | 16.82 Milliards de dollars |
| Taux de croissance (2026 - 2031) | 7.14% CAGR |
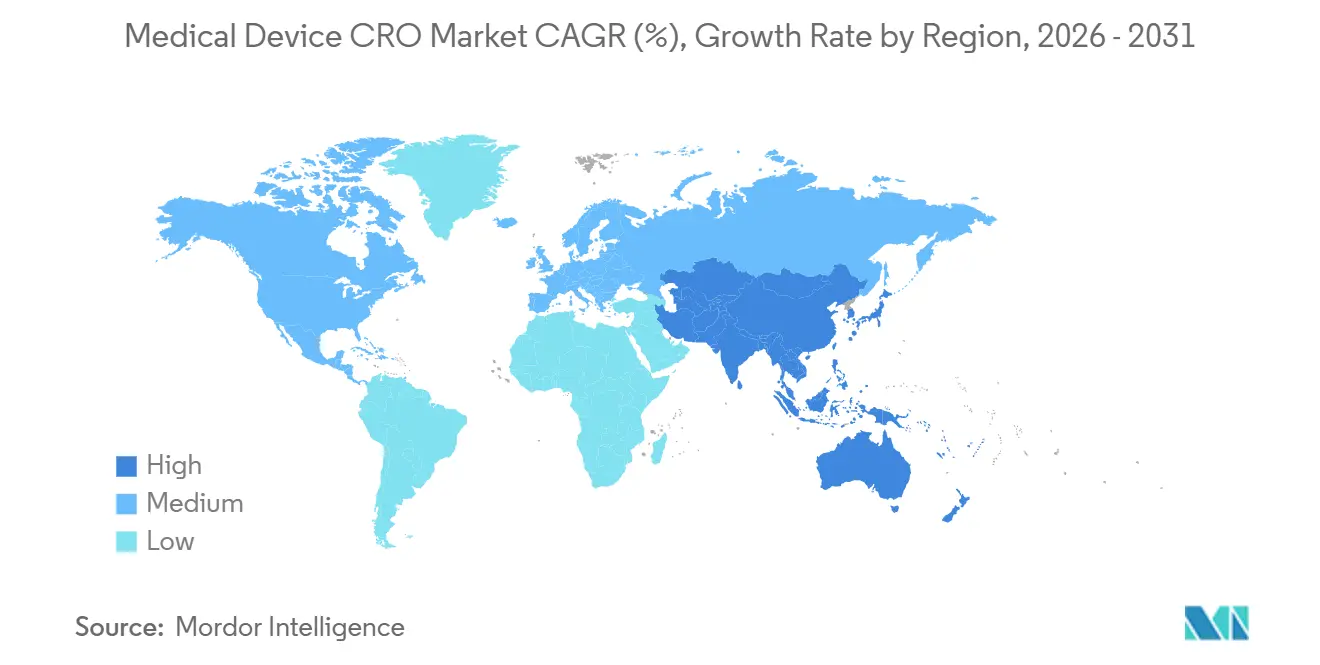
| Marché à la Croissance la Plus Rapide | Asie-Pacifique |
| Plus Grand Marché | Amérique du Nord |
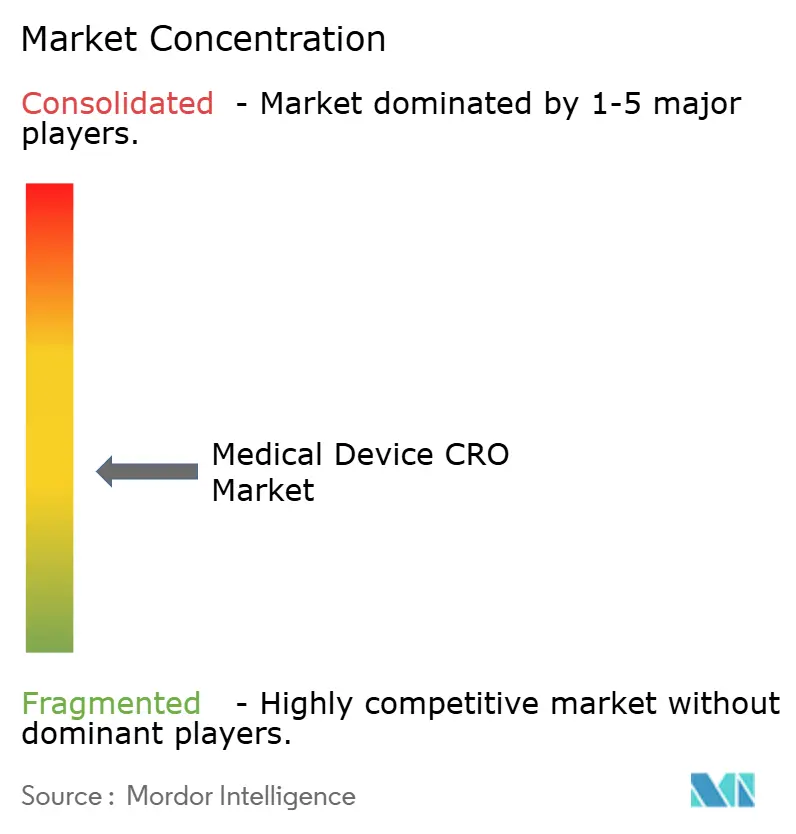
| Concentration du Marché | Moyen |
Acteurs majeurs *Avis de non-responsabilité : les principaux acteurs sont triés sans ordre particulier Image © Mordor Intelligence. La réutilisation nécessite une attribution sous CC BY 4.0. | |
Analyse du marché des CRO de dispositifs médicaux par Mordor Intelligence
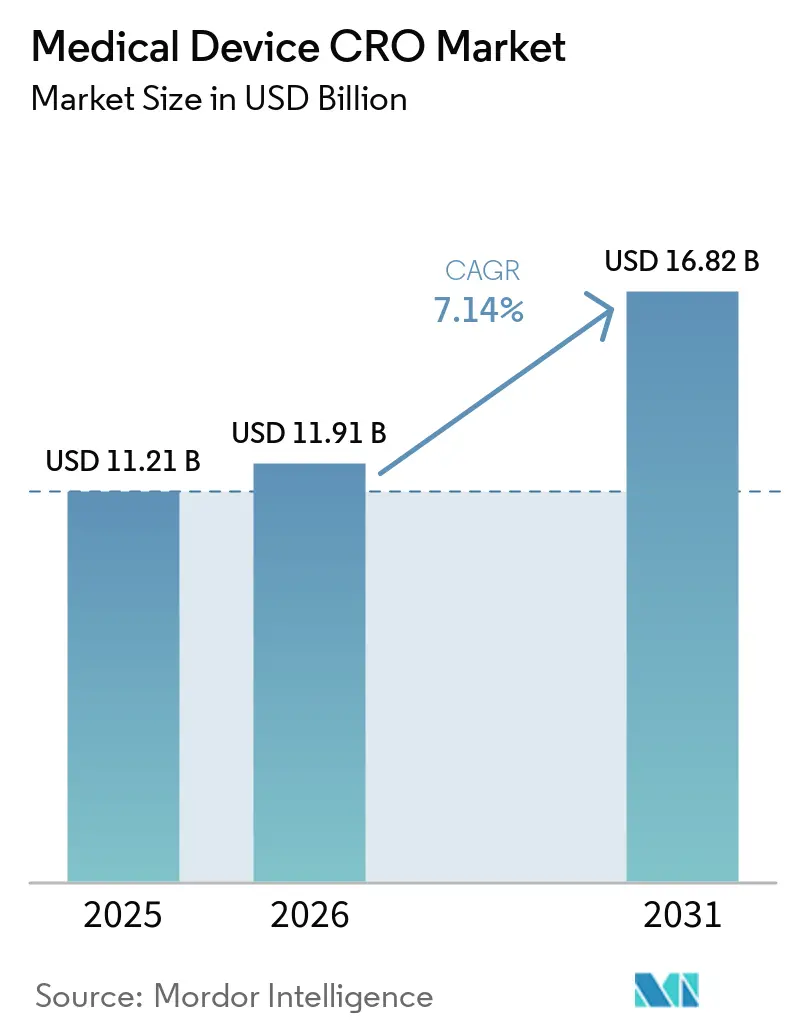
La taille du marché des CRO de dispositifs médicaux devrait augmenter de 11,21 milliards USD en 2025 à 11,91 milliards USD en 2026 et atteindre 16,82 milliards USD d'ici 2031, avec un TCAC de 7,14 % sur la période 2026-2031.
Le marché des CRO de dispositifs médicaux se distingue de l'externalisation pharmaceutique car les commanditaires de dispositifs ont souvent besoin de validation matérielle, de vérification logicielle, de travaux d'utilisabilité et de tests de biocompatibilité dans le même dossier de soumission, ce qui rend les besoins d'externalisation plus fragmentés et plus exigeants sur le plan technique. Le passage au QMSR de la FDA en février 2026 et la charge continue du RDM et du RDIV de l'UE poussent les commanditaires vers des partenaires spécialisés capables de relier l'ingénierie, l'exécution clinique et le travail réglementaire au sein d'un même programme plutôt que dans des transferts séparés. Le marché des CRO de dispositifs médicaux reste également engagé sur des périodes plus longues car le développement de dispositifs est itératif, et les mises à jour de conception déclenchent souvent de nouveaux cycles de documentation, de tests et de revalidation au lieu de suivre un modèle propre phase par phase. Les exigences de gouvernance des données transfrontalières et les goulots d'étranglement de certification en Europe ralentissent encore les délais pour les flux de travail multinationaux, en particulier lorsque les commanditaires ont besoin de dossiers de preuves harmonisés entre les régions. La pression concurrentielle reste modérée à élevée sur le marché des CRO de dispositifs médicaux, et la consolidation récente par de grands prestataires de tests et de services réduit l'espace disponible pour les entreprises de taille intermédiaire qui ne peuvent pas offrir une prestation intégrée sur l'ensemble du cycle de vie.
Principaux enseignements du rapport
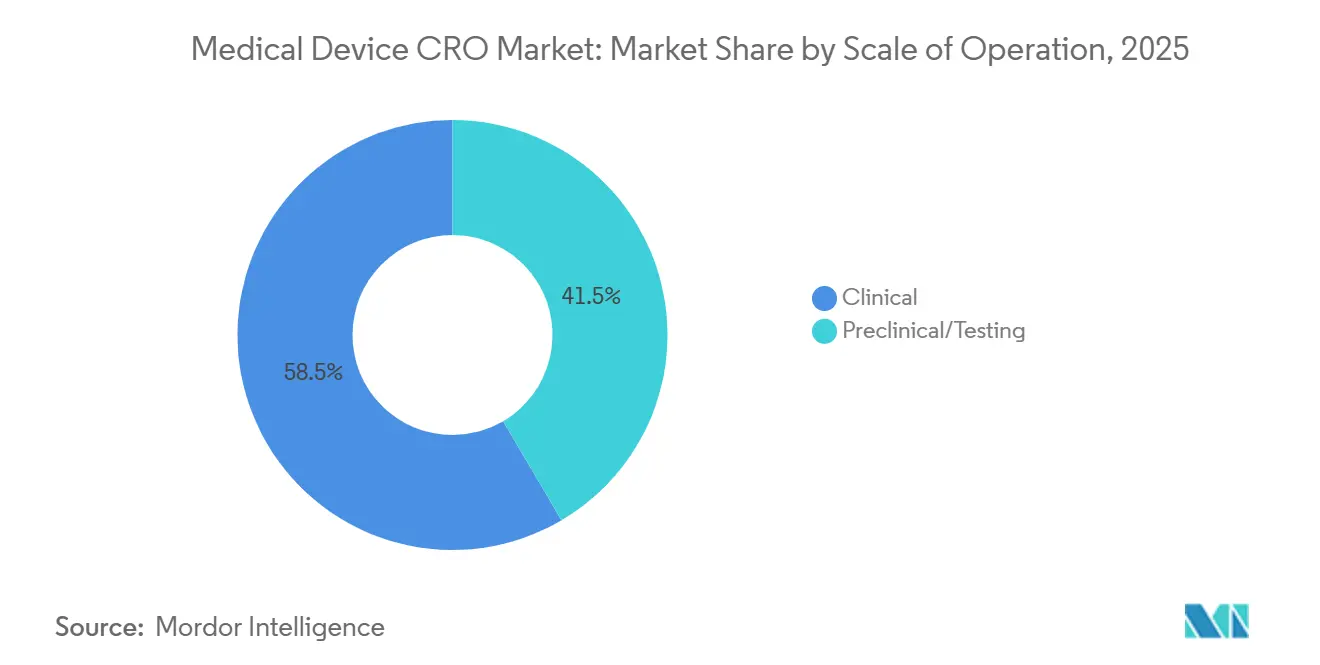
- Par échelle d'opération, les services cliniques détenaient 58,46 % du marché en 2025, et ce même sous-segment devrait se développer à un TCAC de 7,67 % jusqu'en 2031.
- Par type de service, la surveillance clinique était en tête avec une part de 21,29 % en 2025, tandis que les affaires réglementaires et médicales devraient connaître la croissance la plus rapide avec un TCAC de 7,82 % jusqu'en 2031.
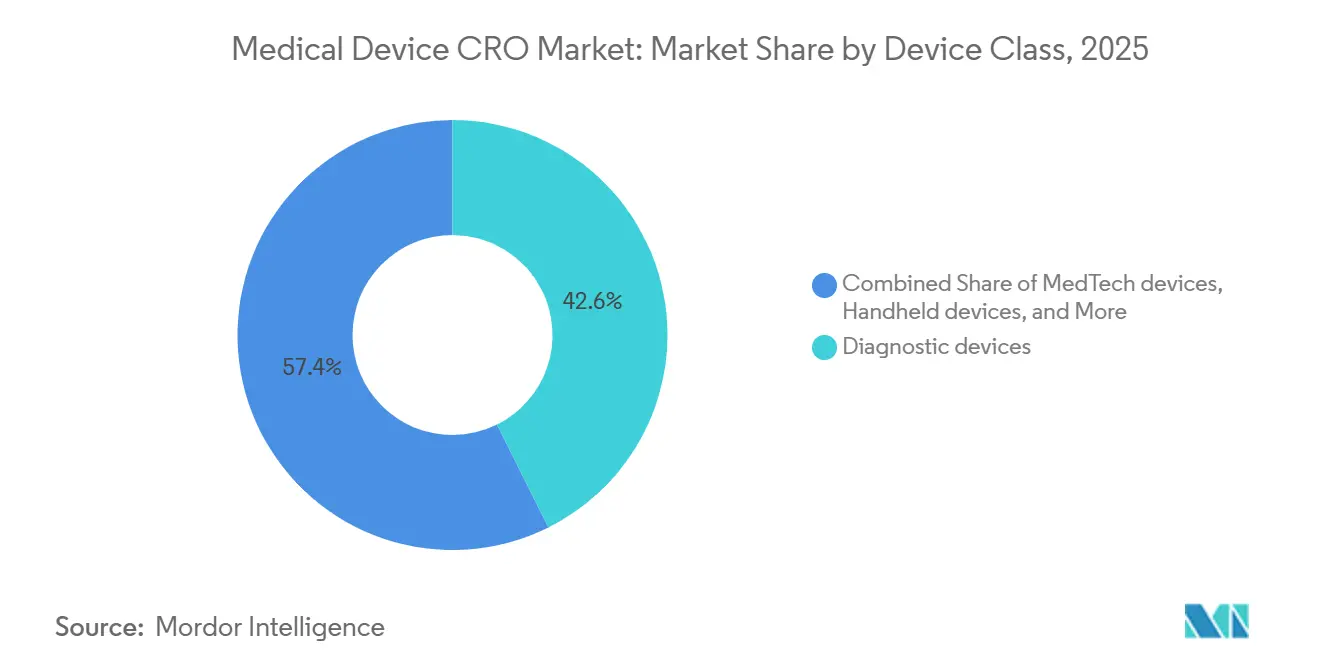
- Par classe de dispositif, les dispositifs de diagnostic représentaient une part de 42,64 % en 2025, tandis que les dispositifs MedTech devraient enregistrer le TCAC le plus élevé à 7,42 % jusqu'en 2031.
- Par géographie, l'Amérique du Nord détenait 45,34 % du marché en 2025, tandis que l'Asie-Pacifique devrait afficher le TCAC régional le plus rapide à 8,32 % jusqu'en 2031.
Note : La taille du marché et les prévisions figurant dans ce rapport sont générées à l'aide du cadre d'estimation exclusif de Mordor Intelligence, mis à jour avec les dernières données et informations disponibles en janvier 2026.
Tendances et perspectives du marché mondial des CRO de dispositifs médicaux
Analyse de l'impact des moteurs*
| Moteur | (~) % d'impact sur les prévisions de TCAC | Pertinence géographique | Horizon temporel de l'impact |
|---|---|---|---|
| L'intensification de l'externalisation à mesure que les essais sur dispositifs augmentent en volume et en complexité | +2.0% | Mondial, plus aigu en Amérique du Nord et en Europe occidentale | Court terme (≤ 2 ans) |
| Les exigences en matière de preuves RDM/RDIV de l'UE et les exigences de SCAP élargissent la demande | +1.8% | Europe en premier lieu, répercussions sur l'APAC et l'Amérique du Nord pour les enregistrements mondiaux | Moyen terme (2-4 ans) |
| Les exigences de cybersécurité de la FDA et l'alignement sur le QMSR élèvent les besoins en validation logicielle | +0.9% | Amérique du Nord en premier lieu, suivi de l'UE et de l'APAC | Moyen terme (2-4 ans) |
| L'adoption des essais décentralisés dans les dispositifs accroît l'externalisation des THN et des solutions eCliniques | +1.2% | Amérique du Nord et Europe en tête, adoption précoce en APAC | Court terme (≤ 2 ans) à moyen terme (2-4 ans) |
| Les contraintes de transfert de données transfrontalières stimulent les services d'ingénierie de la confidentialité | +0.7% | UE, avec pertinence pour les programmes transfrontaliers Amérique du Nord-UE | Long terme (≥ 4 ans) |
| Source: Mordor Intelligence | |||
L'intensification de l'externalisation à mesure que les essais sur dispositifs augmentent en volume et en complexité
Le marché des CRO de dispositifs médicaux bénéficie d'un modèle de preuves cliniques qui s'étend désormais bien au-delà de la soumission préalable à la mise sur le marché, car la surveillance après commercialisation et le SCAP constituent des obligations continues plutôt que des activités ponctuelles. Le MDCG 2025-10 stipule que le SPM doit activement collecter et analyser des données tout au long de la durée de vie du dispositif, et que ces informations doivent alimenter les processus de gestion des risques, d'évaluation clinique, de documentation technique et d'actions correctives. Cette exigence rend plus difficile pour les fabricants de maintenir toutes les tâches réglementaires, biostatistiques, de surveillance et de reporting au sein de leurs propres équipes, en particulier lorsque les produits à risque plus élevé nécessitent également des rapports structurés et des mises à jour continues.
Le marché des CRO de dispositifs médicaux bénéficie donc de travaux récurrents sur les produits existants ainsi que sur les nouveaux lancements, car les anciens dispositifs intégrant de nouveaux cadres réglementaires peuvent rouvrir des lacunes en matière de preuves et déclencher de nouveaux mandats d'externalisation. Medpace a déclaré un chiffre d'affaires annuel 2025 de 2 530,2 millions USD, en hausse de 20 %, et a prévu entre 2,755 milliards USD et 2,855 milliards USD pour 2026, ce qui conforte l'idée que la demande d'externalisation est restée saine dans l'ensemble de la base de services de développement[1]Groupe de coordination des dispositifs médicaux, « Orientations MDCG 2025-10 sur la surveillance après commercialisation des dispositifs médicaux et des dispositifs médicaux de diagnostic in vitro », Commission européenne, health.ec.europa.eu.
Exigences en matière de preuves RDM/RDIV de l'UE et de SCAP élargissant la demande
Le MDCG 2025-10 fait du système de SPM une partie intégrante du système de management de la qualité, ce qui signifie que le travail sur les preuves cliniques s'inscrit désormais dans une boucle de conformité continue plutôt que dans un exercice de dépôt autonome. Les orientations précisent également qu'un plan de SCAP ou de SCAP pour les performances doit être inclus dans le plan de SPM, ou que le fabricant doit justifier pourquoi cette activité n'est pas applicable. Les résultats du SCAP et du SCAP pour les performances doivent ensuite mettre à jour l'évaluation clinique ou des performances, l'évaluation bénéfice-risque, l'étiquetage, la documentation technique et les autres dossiers de conformité en cours. Sur le marché des CRO de dispositifs médicaux, cette structure accroît la demande de surveillance, de gestion des données, de rédaction médicale, de soutien à la vigilance et de maintenance réglementaire, car les commanditaires doivent maintenir les preuves à jour après le lancement et pas seulement avant l'approbation. Les prestataires capables de combiner des capacités de données en vie réelle avec des flux de travail documentaires sont mieux positionnés à mesure que les commanditaires transfèrent davantage de ce travail en dehors des équipes internes déjà sollicitées par les obligations du RDM et du RDIV.
Les exigences de cybersécurité de la FDA et l'alignement sur le QMSR élèvent les besoins en validation logicielle et en tests de cybersécurité
Les orientations de la FDA de février 2026 sur la cybersécurité fournissent des recommandations sur la conception, l'étiquetage et le contenu des soumissions pour les dispositifs présentant un risque de cybersécurité, et elles traitent des attentes de la section 524B pour les cyber-dispositifs. Ce document a remplacé les orientations de juin 2025, ce qui signale que les attentes en matière de cybersécurité évoluent vers une norme opérationnelle plus formelle et actualisée pour les soumissions de dispositifs. Parallèlement, le QMSR est entré en vigueur le 2 février 2026 et a intégré la norme ISO 13485:2016, tout en mettant l'accent sur la gestion des risques tout au long des processus de conception, de développement, de production et d'autres processus du cycle de vie. La règle du Registre fédéral a également ajouté des exigences détaillées en matière de dossiers de réclamations, de dossiers d'entretien et de documentation liée à l'IUD qui alourdissent la charge de travail pour les programmes de dispositifs connectés et riches en logiciels. Cette combinaison élargit le rôle des fournisseurs spécialisés en validation logicielle et en tests de cybersécurité sur le marché des CRO de dispositifs médicaux, car de nombreux commanditaires ne disposent pas des outils internes ou du personnel nécessaires pour maintenir ces flux de travail à jour à grande échelle[2]Food and Drug Administration, « Dispositifs médicaux, amendements à la réglementation sur les systèmes de qualité », Registre fédéral, govinfo.gov.
L'adoption des essais décentralisés dans les dispositifs accroît l'externalisation des THN/solutions eCliniques et des opérations à distance
L'adoption des essais décentralisés devient de plus en plus pertinente pour le marché des CRO de dispositifs médicaux car les dispositifs représentaient 21 % des enregistrements d'essais cliniques décentralisés interventionnels dans une revue mondiale de 1 370 études. La même analyse a montré que seule une petite minorité d'ECD fonctionnait sans outils numériques, ce qui confirme que l'exécution à distance des essais dépend désormais des systèmes de données, des outils de communication et des flux de travail numériques. Dans le même temps, l'activité des ECD est restée concentrée dans les pays à revenus élevés, ce qui suggère que les modèles à distance s'étendent au sein des systèmes de recherche établis avant d'élargir pleinement l'accès à de nouveaux bassins de patients. Ce schéma soutient néanmoins la demande d'externalisation, car les commanditaires ont besoin d'aide pour les systèmes eCliniques, la coordination à distance des sites, la gestion des données distribuées et les méthodes de surveillance hybrides dans plusieurs zones géographiques. Le marché des CRO de dispositifs médicaux bénéficie donc de l'adoption des ECD, mais les gagnants seront probablement les prestataires capables de combiner les opérations d'essais numériques avec les besoins de gestion pratique que les études sur dispositifs requièrent encore.
Analyse de l'impact des freins*
| Frein | (~) % d'impact sur les prévisions de TCAC | Pertinence géographique | Horizon temporel de l'impact |
|---|---|---|---|
| Les goulots d'étranglement de capacité des organismes notifiés allongent les certifications dans l'UE | -0.8% | Europe en premier lieu, impact secondaire sur les doubles soumissions Amérique du Nord-UE | Court terme (≤ 2 ans) à moyen terme (2-4 ans) |
| Les obstacles au recrutement de patients pour les ECD, à l'accès technologique et à la gouvernance des données ralentissent l'exécution | -0.5% | Mondial, plus prononcé dans les marchés émergents à faibles revenus et en APAC | Moyen terme (2-4 ans) |
| Les obligations de SBOM de cybersécurité et de correction ajoutent une charge de revalidation récurrente | -0.4% | Amérique du Nord en premier lieu, facteur de conformité émergent dans l'UE | Long terme (≥ 4 ans) |
| Source: Mordor Intelligence | |||
Les goulots d'étranglement de capacité des organismes notifiés allongent les certifications dans l'UE
La capacité des organismes notifiés reste l'un des risques de délai les plus évidents pour le marché des CRO de dispositifs médicaux en Europe, car le flux de certifications accuse encore un retard par rapport au volume de travail transitant par les voies RDM et RDIV. MedTech Europe a décrit les organismes notifiés comme un goulot d'étranglement réglementaire, et cette formulation s'aligne sur la pression soutenue exercée sur les commanditaires qui ont besoin d'approbations, de renouvellements et de mises à jour des preuves dans le cadre de règles plus strictes. Lorsque les délais de certification glissent, les contrats liés à des jalons peuvent retarder la comptabilisation des revenus pour les CRO même si le travail clinique ou documentaire a déjà été achevé. Cela ne supprime pas la demande sur le marché des CRO de dispositifs médicaux, mais rend les calendriers de livraison européens plus difficiles à prévoir et accroît la valeur des prestataires capables de combiner le travail clinique avec une coordination réglementaire étroite. En pratique, le goulot d'étranglement déplace la concurrence vers les entreprises capables de gérer le risque de retard, de séquencer soigneusement les flux de travail et de maintenir la confiance des commanditaires pendant de longs cycles d'examen[3]MedTech Europe, « Les organismes notifiés deviennent un goulot d'étranglement réglementaire », MedTech Europe, medtecheurope.org.
Les obstacles au recrutement de patients pour les ECD, à l'accès technologique et à la gouvernance des données ralentissent l'exécution des études
Les modèles décentralisés peuvent étendre la portée des essais, mais les données montrent encore que la plupart des activités d'ECD dans un seul pays sont concentrées dans des contextes à revenus élevés plutôt que dans des bassins de patients mal desservis. Cette concentration signifie que les avantages de l'exécution à distance sont inégaux, en particulier lorsque les commanditaires s'attendent à des gains rapides d'enrôlement sur des marchés présentant des capacités d'accès numérique et de sites très différentes. La Commission européenne a précisé que le règlement sur les essais cliniques et le RGPD s'appliquent simultanément, ce qui ajoute des obligations en matière de base juridique, de stockage, d'archivage, de droits des sujets, de mesures de sécurité et de transferts internationaux de données. Le CEPD a également traité cette interaction comme un problème de conformité distinct, ce qui renforce la façon dont la gouvernance des données peut ralentir le démarrage des études lorsque plusieurs parties, systèmes et pays sont impliqués. Pour le marché des CRO de dispositifs médicaux, cela signifie que l'exécution des ECD nécessite encore plus de soutien juridique, opérationnel et technique que de nombreux commanditaires ne l'avaient d'abord supposé, ce qui réduit l'avantage de rapidité que les modèles à distance peuvent par ailleurs offrir.
*Nos prévisions considèrent les impacts des moteurs et des contraintes comme directionnels et non additifs. Les prévisions d'impact reflètent la croissance de référence, les effets de composition et les interactions entre variables.
Analyse des segments
Par échelle d'opération : les services cliniques définissent le programme d'externalisation
Les services cliniques détenaient 58,46 % de la part du marché des CRO de dispositifs médicaux en 2025, et ce même segment devrait croître à un TCAC de 7,67 % jusqu'en 2031. Cette position de leader reflète plus que le simple volume d'essais, car les fabricants font désormais face à des obligations cliniques qui se poursuivent dans la période post-commercialisation et alimentent la gestion des risques et la documentation technique. Le MDCG 2025-10 rend cette continuité explicite en traitant le SPM comme un système actif et continu qui doit mettre à jour l'évaluation bénéfice-risque, l'évaluation clinique et les actions correctives tout au long de la durée de vie du dispositif. Pour le marché des CRO de dispositifs médicaux, cela signifie que le travail clinique n'est plus concentré uniquement autour de la première soumission, car le SCAP et la maintenance des preuves associées prolongent l'activité bien après le lancement. Il en résulte un bassin d'externalisation plus large et plus durable pour les commanditaires disposant de produits de classe IIb, classe III, implantables et riches en logiciels qui ne peuvent pas assurer ces tâches en interne au rythme requis.
Les services précliniques et de tests restent plus modestes au sein du marché des CRO de dispositifs médicaux, mais ils deviennent plus stratégiquement importants car ils se situent en amont de nombreuses décisions réglementaires et cliniques ultérieures. Les commanditaires de dispositifs ont encore besoin de plans d'évaluation biologique, de caractérisation chimique, d'évaluations toxicologiques et d'analyses des lacunes qui s'alignent sur les attentes réglementaires actuelles plutôt que sur d'anciennes hypothèses d'approbation. L'offre de biocompatibilité déclarée de Labcorp montre comment ces services couvrent déjà les tests ISO 10993, la caractérisation chimique, l'évaluation des risques toxicologiques et le soutien aux examens des lacunes des dispositifs existants liés aux normes réglementaires actuelles. Cela maintient le secteur des CRO de dispositifs médicaux dépendant des prestataires de tests capables de relier le travail de laboratoire à la stratégie de soumission, plutôt que d'agir uniquement comme des laboratoires isolés au début du développement.


Par type de service : les affaires réglementaires et médicales dépassent toutes les autres lignes de services
La surveillance clinique a conservé une part de 21,29 % de la taille du marché des CRO de dispositifs médicaux en 2025, tandis que les affaires réglementaires et médicales devraient se développer à un TCAC de 7,82 % jusqu'en 2031. Ce schéma de croissance reflète l'ampleur des changements réglementaires actuels aux États-Unis et en Europe, où les commanditaires ont désormais besoin d'un soutien continu accru en matière de documentation, d'alignement qualité, de soumissions et de maintenance post-commercialisation. Le QMSR est entré en vigueur en 2026 et a intégré la norme ISO 13485:2016 dans le 21 CFR Partie 820, tout en préservant les contrôles d'enregistrement spécifiques à la FDA tels que les dossiers de réclamations, les dossiers d'entretien et la documentation liée à l'IUD. Ces exigences rendent le travail réglementaire plus continu, car les commanditaires doivent maintenir des systèmes et des dossiers conformes tout au long de la vie du produit plutôt que d'assembler un dossier de soumission restreint à un moment donné. Sur le marché des CRO de dispositifs médicaux, cela transforme les affaires réglementaires et médicales en moteur de croissance pour les prestataires capables de combiner qualité, étiquetage, documentation et soutien face aux régulateurs dans un seul modèle opérationnel.
La surveillance clinique sous-tend encore les revenus sur le marché des CRO de dispositifs médicaux car chaque évolution réglementaire plus large dépend encore d'une supervision fiable des sites, d'un flux de données et d'une escalade des problèmes pendant les études actives. L'utilisation croissante des outils numériques et des modèles à distance modifie la façon dont la surveillance est assurée, mais elle n'a pas réduit le besoin d'un contrôle opérationnel rigoureux dans les programmes de dispositifs. La gestion des données et biostatistiques, la rédaction médicale et le recrutement des patients et des sites continuent d'agir comme des fonctions habilitantes qui rendent possible la prestation clinique et réglementaire à grande échelle. Au sein du secteur des CRO de dispositifs médicaux, les prestataires de services qui intègrent étroitement ces fonctions sont mieux positionnés que les entreprises qui vendent chaque flux de travail comme une tâche distincte avec des équipes et des calendriers séparés.
Par classe de dispositif : les dispositifs de diagnostic sont en tête, mais les dispositifs MedTech s'accélèrent
Les dispositifs de diagnostic représentaient 42,64 % du marché en 2025, ce qui les maintenait comme la plus grande classe de dispositifs au sein du marché des CRO de dispositifs médicaux. Leur avance est étroitement liée à la pression du RDIV, car une part bien plus importante des diagnostics in vitro nécessite désormais l'intervention d'un organisme notifié que sous l'ancienne directive. Ce changement augmente la quantité de preuves de performance, de travail documentaire et de préparation du système qualité que les fabricants doivent réaliser avant et après l'accès au marché. Le marché des CRO de dispositifs médicaux constate donc une forte demande en matière de diagnostics non seulement pour les nouveaux produits, mais aussi pour les produits transitant par des voies de reclassification et de transition qui créent un travail de preuve supplémentaire. C'est l'une des raisons pour lesquelles les diagnostics continuent de générer une demande d'externalisation régulière même lorsque les délais de certification restent inégaux en Europe.
Les dispositifs MedTech devraient connaître la croissance la plus rapide avec un TCAC de 7,42 % jusqu'en 2031, ce qui reflète une activité croissante autour des systèmes connectés, des produits pilotés par logiciel et des plateformes implantables complexes. Les attentes actuelles de la FDA en matière de cybersécurité et de SMQ augmentent la quantité de validation, de documentation et de gestion des risques sur le cycle de vie requise pour ces produits avant que les commanditaires puissent progresser efficacement dans l'examen et la maintenance post-commercialisation. Les produits portables et à usage domestique ajoutent également du travail sur le marché des CRO de dispositifs médicaux car ils nécessitent souvent des dossiers de preuves reliant les conditions d'utilisation réelles aux attentes réglementaires et qualité. Au sein du secteur des CRO de dispositifs médicaux, le mix évolue donc vers des prestataires capables de gérer la validation logicielle, la sécurité biologique, les preuves cliniques et la rédaction réglementaire sans obliger les commanditaires à coordonner plusieurs spécialistes déconnectés.

Analyse géographique
L'Amérique du Nord détenait 45,34 % de la part du marché des CRO de dispositifs médicaux en 2025, ce qui en faisait le plus grand contributeur régional. Les États-Unis représentent la majeure partie de cette position car le calendrier réglementaire a concentré plusieurs exigences de conformité sur une courte période, notamment à travers la mise en œuvre du QMSR et les attentes actualisées en matière de cybersécurité. Le QMSR intègre désormais la norme ISO 13485:2016 par référence, et il maintient également en place les contrôles d'enregistrement spécifiques à la FDA, ce qui signifie que les fabricants doivent gérer l'harmonisation sans supposer que la certification ISO seule remplace l'inspection ou les obligations documentaires de la FDA. De nombreux commanditaires comblent cette lacune par l'externalisation plutôt qu'en s'appuyant uniquement sur le recrutement interne en assurance qualité et affaires réglementaires, ce qui contribue à soutenir le marché des CRO de dispositifs médicaux en Amérique du Nord même dans un environnement de dépenses plus discipliné. Cela fait de la région l'exemple le plus clair de la façon dont la réglementation peut élargir la demande d'externalisation par la complexité de la conformité plutôt que par le seul volume d'essais.
L'Europe reste le deuxième pôle régional du marché des CRO de dispositifs médicaux, et c'est la géographie où la réglementation remodèle le plus directement le mix de services. Le RDM et le RDIV ont intégré davantage de preuves post-commercialisation, de maintenance documentaire et d'interaction avec les organismes notifiés dans le modèle opérationnel, ce qui accroît la dépendance envers les CRO capables de combiner travail clinique et réglementaire. Dans le même temps, les goulots d'étranglement des organismes notifiés continuent de compliquer les délais, ce qui signifie que la demande de CRO reste forte même si la conversion des contrats et le rythme des jalons deviennent plus difficiles à prévoir. Cela laisse l'Europe comme une région avec une force de demande structurelle sur le marché des CRO de dispositifs médicaux, mais aussi avec un risque de délai de revenus plus visible qu'en Amérique du Nord.
L'Asie-Pacifique devrait enregistrer le TCAC le plus rapide à 8,32 % sur la taille du marché des CRO de dispositifs médicaux jusqu'en 2031. La région bénéficie d'une activité croissante de développement de dispositifs, mais les voies réglementaires locales rendent les capacités sur le terrain importantes plutôt qu'optionnelles. La PMDA maintient des structures formelles de notification et de consultation pour les essais cliniques sur les dispositifs médicaux, et ces processus renforcent la nécessité d'une gestion réglementaire localisée plutôt que d'un modèle mondial unique. Cela soutient la demande sur le marché des CRO de dispositifs médicaux pour des prestataires capables de combiner accès régional aux sites, soutien linguistique et exécution face aux régulateurs au Japon et sur les marchés voisins. La Chine et l'Inde élargissent également la base de demande future à mesure que les pipelines de fabrication nationaux créent davantage de travail clinique et réglementaire que les commanditaires peuvent préférer externaliser. La Corée du Sud ajoute à l'attrait régional car un environnement d'approbation plus efficace peut soutenir des modèles d'essais hybrides et des empreintes d'études régionales plus larges. Le Moyen-Orient et l'Afrique, ainsi que l'Amérique du Sud, restent plus modestes sur le marché des CRO de dispositifs médicaux, mais les programmes d'enregistrement multinationaux dans les pays du CCG, au Brésil et en Argentine créent encore des opportunités pour les prestataires internationaux disposant d'une portée opérationnelle locale.

Paysage concurrentiel
Le marché des CRO de dispositifs médicaux est modérément fragmenté, avec un nombre limité d'organisations mondiales à service complet et un champ beaucoup plus large d'entreprises spécialisées axées sur les tests, les affaires réglementaires ou la validation numérique. Cette structure est importante car les commanditaires de dispositifs ont souvent besoin de plusieurs disciplines à la fois, et tous les prestataires ne peuvent pas relier sécurité biologique, opérations cliniques, validation logicielle et soutien aux soumissions dans un seul engagement. La consolidation devient donc un outil concurrentiel pratique sur le marché des CRO de dispositifs médicaux, en particulier pour les entreprises cherchant à passer d'ensembles de capacités étroites vers une prestation de bout en bout. L'acquisition par NAMSA des opérations de tests de dispositifs médicaux américains de WuXi AppTec en février 2025 a élargi la capacité de laboratoire et l'étendue des services, tandis que son accord de janvier 2026 pour l'activité de tests de dispositifs médicaux en développement précoce de Labcorp a renforcé la profondeur des tests en microbiologie, biocompatibilité et domaines connexes. NAMSA indique également que plus de 70 % des études mondiales de biocompatibilité sont menées par son organisation, ce qui montre à quel point certaines niches critiques pour la conformité sont déjà concentrées même si le marché des CRO de dispositifs médicaux dans son ensemble reste plus dispersé.
D'autres grands prestataires élargissent également leur portée par des mouvements de portefeuille ciblés plutôt qu'en s'appuyant uniquement sur la croissance organique. ICON a divulgué son acquisition de KCR S.A. Group pour 92,5 millions USD en 2024, et en décembre 2025, il a annoncé un accord pour acquérir ClinicalRM afin de renforcer sa position dans la recherche financée par les gouvernements et les travaux sur les maladies infectieuses pouvant également soutenir les programmes de dispositifs. L'intégration complète en mars 2025 de Medidata Clinical Data Studio par ICON pointe également vers une voie de différenciation basée sur la technologie, car la plateforme rassemble des données provenant de sources Medidata et non-Medidata dans un flux de travail unique pour la gestion des données, l'examen et la surveillance centralisée. Ces étapes montrent que le marché des CRO de dispositifs médicaux ne se concurrence pas uniquement sur les effectifs ou la portée géographique, car la profondeur de la plateforme et l'intégration des flux de travail font partie de l'offre commerciale.
La technologie devient une frontière concurrentielle de plus en plus visible sur le marché des CRO de dispositifs médicaux car les commanditaires souhaitent de plus en plus un partenaire unique capable de travailler sur l'examen des données, la surveillance, les dossiers qualité et la documentation réglementaire sans transferts répétés. Dans le même temps, les prestataires spécialisés restent importants car le travail sur les dispositifs comprend des catégories étroites mais essentielles telles que la biocompatibilité, les extractibles et les lixiviables, et la validation axée sur la cybersécurité que les grandes entreprises généralistes ne contrôlent pas toujours en interne. Le marché connaît donc deux voies simultanément, avec de grandes entreprises qui s'étendent par acquisition et intégration tandis que les entreprises de niche défendent leur position par la profondeur technique et une réponse plus rapide dans des lignes de services ciblées. L'espace blanc reste le plus visible là où les dispositifs riches en logiciels nécessitent un soutien aligné sur les preuves cliniques, la gestion des données respectueuse de la vie privée et la maintenance qualité sur le cycle de vie. Cela maintient le marché des CRO de dispositifs médicaux compétitif, actif et seulement modérément concentré malgré le rôle plus fort de quelques acteurs à grande échelle dans certaines niches de tests.
Leaders du secteur des CRO de dispositifs médicaux
IQVIA
NAMSA
Medpace
Charles River Laboratories
Avania
- *Avis de non-responsabilité : les principaux acteurs sont triés sans ordre particulier

Développements récents du secteur
- Février 2026 : le QMSR de la FDA (21 CFR Partie 820 tel qu'amendé) est entré en vigueur le 2 février, intégrant la norme ISO 13485:2016 par référence, retirant la technique d'inspection du système qualité (QSIT) au profit d'un processus d'inspection mis à jour dans le cadre du programme de conformité 7382.850. Le QMSR introduit une intégration obligatoire de la gestion des risques tout au long du cycle de vie du produit et de nouvelles exigences de tenue de dossiers liées à l'IUD, élargissant la demande de CRO pour les services d'évaluation des lacunes du SMQ et de revalidation.
- Janvier 2026 : Labcorp a annoncé la vente de son activité de tests de dispositifs médicaux en développement précoce à NAMSA. La transaction élargit la capacité de tests de NAMSA en microbiologie, biocompatibilité et extractibles/lixiviables, renforçant sa position de CRO de tests précliniques dominant dans le secteur des dispositifs médicaux.


Périmètre du rapport sur le marché mondial des CRO de dispositifs médicaux
Selon le périmètre du rapport, une organisation de recherche contractuelle (CRO) de dispositifs médicaux est une entreprise spécialisée qui fournit des services externalisés aux fabricants de dispositifs médicaux. Ces services soutiennent le développement, les tests et l'approbation réglementaire des dispositifs médicaux. Les CRO aident à rationaliser les essais cliniques, les soumissions réglementaires et les activités de conformité, permettant aux fabricants de mettre leurs dispositifs sur le marché plus efficacement.
Le marché des CRO de dispositifs médicaux est segmenté par échelle d'opération en phase préclinique/tests et phase clinique. Par type de service, le marché est catégorisé en services de surveillance clinique, services de gestion des essais cliniques, services de gestion des données et biostatistiques, services d'affaires réglementaires et médicales, services de rédaction médicale, services de recrutement des patients et des sites, services de sécurité et pharmacovigilance, et autres services. Par classe de dispositif, la segmentation comprend les dispositifs MedTech, les dispositifs de diagnostic, les dispositifs portables et les autres dispositifs. Par géographie, le marché est divisé en région Amérique du Nord, région Europe, région Asie-Pacifique, région Moyen-Orient et Afrique, et région Amérique du Sud. Le rapport de marché couvre également les tailles de marché estimées et les tendances pour 17 pays dans les principales régions du monde. Pour chaque segment, la taille du marché et les prévisions sont fournies en termes de valeur (USD).
| Préclinique/Tests |
| Clinique |
| Surveillance clinique |
| Gestion des essais cliniques |
| Gestion des données et biostatistiques |
| Affaires réglementaires et médicales |
| Rédaction médicale |
| Recrutement des patients et des sites |
| Sécurité et pharmacovigilance |
| Autres (activation des ECD et plateformes eCliniques, imagerie/laboratoire central, etc.) |
| Dispositifs MedTech |
| Dispositifs de diagnostic |
| Dispositifs portables |
| Autres |
| Amérique du Nord | États-Unis |
| Canada | |
| Mexique | |
| Europe | Allemagne |
| Royaume-Uni | |
| France | |
| Italie | |
| Espagne | |
| Reste de l'Europe | |
| Asie-Pacifique | Chine |
| Inde | |
| Japon | |
| Corée du Sud | |
| Australie | |
| Reste de l'Asie-Pacifique | |
| Moyen-Orient et Afrique | CCG |
| Afrique du Sud | |
| Reste du Moyen-Orient et de l'Afrique | |
| Amérique du Sud | Brésil |
| Argentine | |
| Reste de l'Amérique du Sud |
| Par échelle d'opération | Préclinique/Tests | |
| Clinique | ||
| Par type de service | Surveillance clinique | |
| Gestion des essais cliniques | ||
| Gestion des données et biostatistiques | ||
| Affaires réglementaires et médicales | ||
| Rédaction médicale | ||
| Recrutement des patients et des sites | ||
| Sécurité et pharmacovigilance | ||
| Autres (activation des ECD et plateformes eCliniques, imagerie/laboratoire central, etc.) | ||
| Par classe de dispositif | Dispositifs MedTech | |
| Dispositifs de diagnostic | ||
| Dispositifs portables | ||
| Autres | ||
| Par géographie | Amérique du Nord | États-Unis |
| Canada | ||
| Mexique | ||
| Europe | Allemagne | |
| Royaume-Uni | ||
| France | ||
| Italie | ||
| Espagne | ||
| Reste de l'Europe | ||
| Asie-Pacifique | Chine | |
| Inde | ||
| Japon | ||
| Corée du Sud | ||
| Australie | ||
| Reste de l'Asie-Pacifique | ||
| Moyen-Orient et Afrique | CCG | |
| Afrique du Sud | ||
| Reste du Moyen-Orient et de l'Afrique | ||
| Amérique du Sud | Brésil | |
| Argentine | ||
| Reste de l'Amérique du Sud | ||


Questions clés auxquelles le rapport répond
Qu'est-ce qui stimule la croissance des services CRO de dispositifs médicaux jusqu'en 2031 ?
La croissance est soutenue par une charge réglementaire croissante, des besoins en preuves post-commercialisation plus longs, et le passage de 11,91 milliards USD en 2026 à 16,82 milliards USD d'ici 2031 à un TCAC de 7,14 %.
Quel domaine de service est en tête de la demande externalisée pour les commanditaires de dispositifs ?
Les services cliniques sont en tête avec une part de 58,46 % en 2025, et constituent également le segment d'échelle d'opération à la croissance la plus rapide avec un TCAC de 7,67 % jusqu'en 2031.
Pourquoi les affaires réglementaires et médicales croissent-elles plus vite que les autres lignes de services ?
Le QMSR, le RDM et le RDIV ont augmenté la quantité de documentation, d'alignement qualité et de travail de conformité sur le cycle de vie, c'est pourquoi les affaires réglementaires et médicales devraient croître à 7,82 % jusqu'en 2031.
Quelle classe de dispositif crée la plus grande opportunité d'externalisation aujourd'hui ?
Les dispositifs de diagnostic détenaient la plus grande part à 42,64 % en 2025 car le RDIV a élargi les besoins en preuves et en documentation pour une plus grande partie de la catégorie.
Quelle région se développe le plus rapidement pour le travail de développement de dispositifs externalisé ?
L'Asie-Pacifique devrait connaître la croissance la plus rapide à 8,32 % jusqu'en 2031, soutenue par une activité de développement régional croissante et la nécessité d'une exécution réglementaire localisée.
Quelle est la concentration de la concurrence parmi les prestataires de CRO de dispositifs médicaux ?
La concurrence est modérée plutôt que très concentrée, car les grandes entreprises mondiales à service complet sont actives, mais de nombreux prestataires spécialisés occupent encore des positions importantes dans les tests, le travail réglementaire et la validation numérique.
Dernière mise à jour de la page le: