Tamanho e Participação do Mercado de CRO de Dispositivos Médicos
Visão Geral do Mercado
| Período de Estudo | 2020 - 2031 |
|---|---|
| Tamanho do Mercado (2026) | 11.91 Bilhões de dólares |
| Tamanho do Mercado (2031) | 16.82 Bilhões de dólares |
| Taxa de crescimento (2026 - 2031) | 7.14% CAGR |
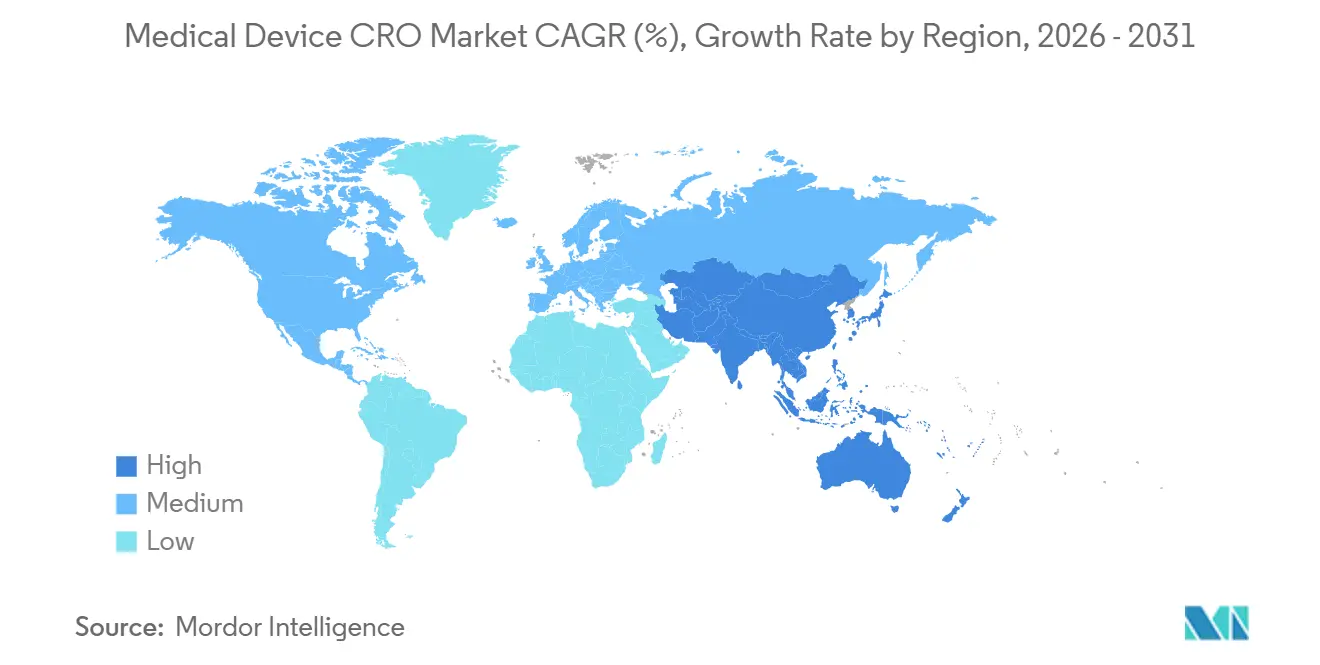
| Mercado de Crescimento Mais Rápido | Ásia-Pacífico |
| Maior Mercado | América do Norte |
| Concentração do Mercado | Médio |
Principais jogadores *Isenção de responsabilidade: Principais participantes classificados em nenhuma ordem específica Imagem © Mordor Intelligence. O reuso requer atribuição conforme CC BY 4.0. | |
Análise do Mercado de CRO de Dispositivos Médicos por Mordor Intelligence
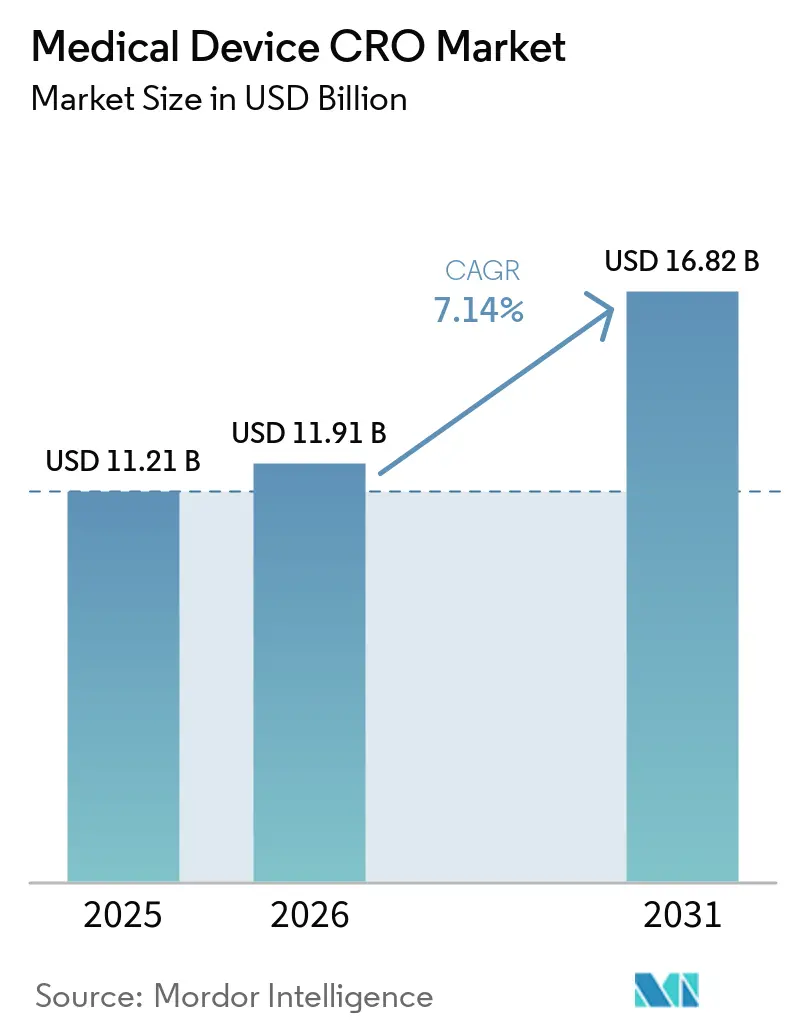
O tamanho do Mercado de CRO de Dispositivos Médicos deve aumentar de USD 11,21 bilhões em 2025 para USD 11,91 bilhões em 2026 e atingir USD 16,82 bilhões até 2031, crescendo a um CAGR de 7,14% no período de 2026 a 2031.
O mercado de CRO de dispositivos médicos difere da terceirização farmacêutica porque os patrocinadores de dispositivos frequentemente precisam de validação de hardware, verificação de software, trabalho de usabilidade e testes de biocompatibilidade dentro do mesmo pacote de submissão, o que torna as necessidades de terceirização mais fragmentadas e mais exigentes do ponto de vista técnico. A transição para o QMSR da FDA em fevereiro de 2026 e o contínuo ônus do MDR e IVDR da UE estão impulsionando os patrocinadores em direção a parceiros especializados que possam conectar engenharia, execução clínica e trabalho regulatório dentro de um único programa, em vez de em transferências separadas. O mercado de CRO de dispositivos médicos também permanece engajado por períodos mais longos porque o desenvolvimento de dispositivos é iterativo, e as atualizações de design frequentemente desencadeiam novas rodadas de documentação, testes e revalidação, em vez de seguir um modelo limpo fase a fase. As exigências de governança de dados transfronteiriços e os gargalos de certificação na Europa ainda retardam o cronograma para fluxos de trabalho multinacionais, especialmente quando os patrocinadores precisam de pacotes de evidências alinhados entre regiões. A pressão competitiva permanece de moderada a alta no mercado de CRO de dispositivos médicos, e a recente consolidação por parte de fornecedores maiores de testes e serviços está estreitando o espaço para empresas de médio porte que não conseguem oferecer entrega integrada ao longo do ciclo de vida.
Principais Conclusões do Relatório
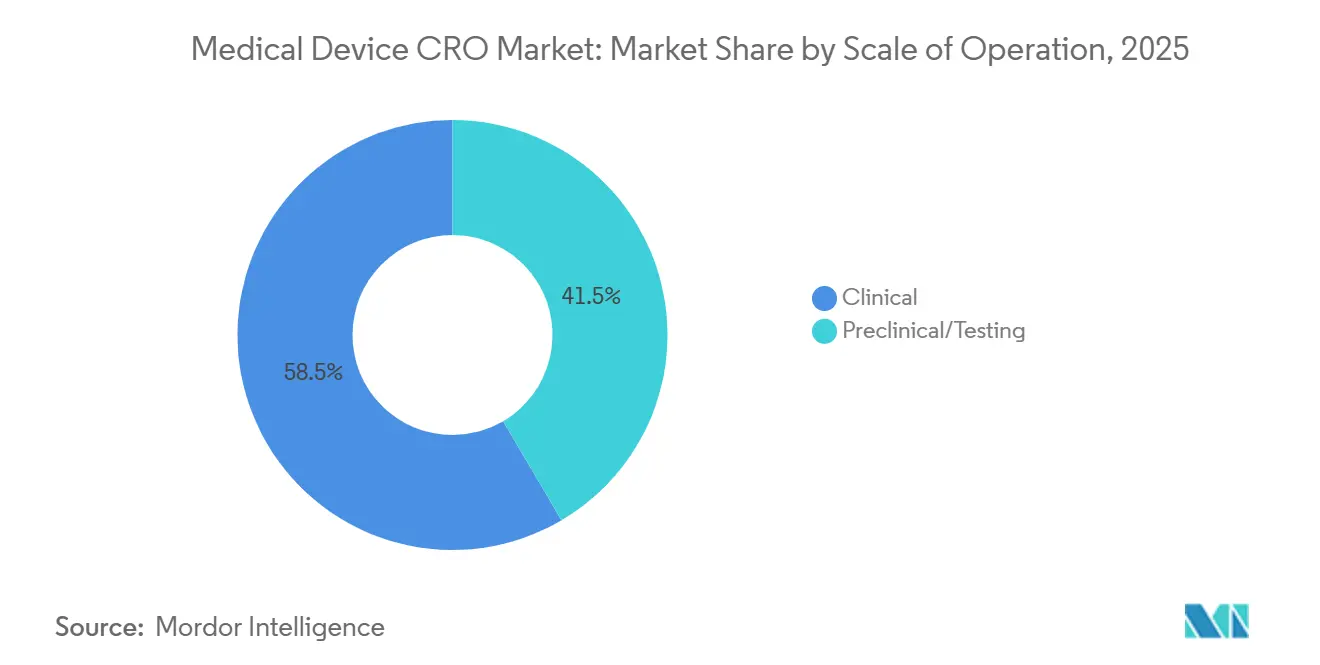
- Por escala de operação, os serviços clínicos detinham 58,46% do mercado em 2025, e o mesmo subsegmento está projetado para expandir a um CAGR de 7,67% até 2031.
- Por tipo de serviço, o Monitoramento Clínico liderou com uma participação de 21,29% em 2025, enquanto Assuntos Regulatórios e Médicos está previsto para crescer mais rapidamente a um CAGR de 7,82% até 2031.
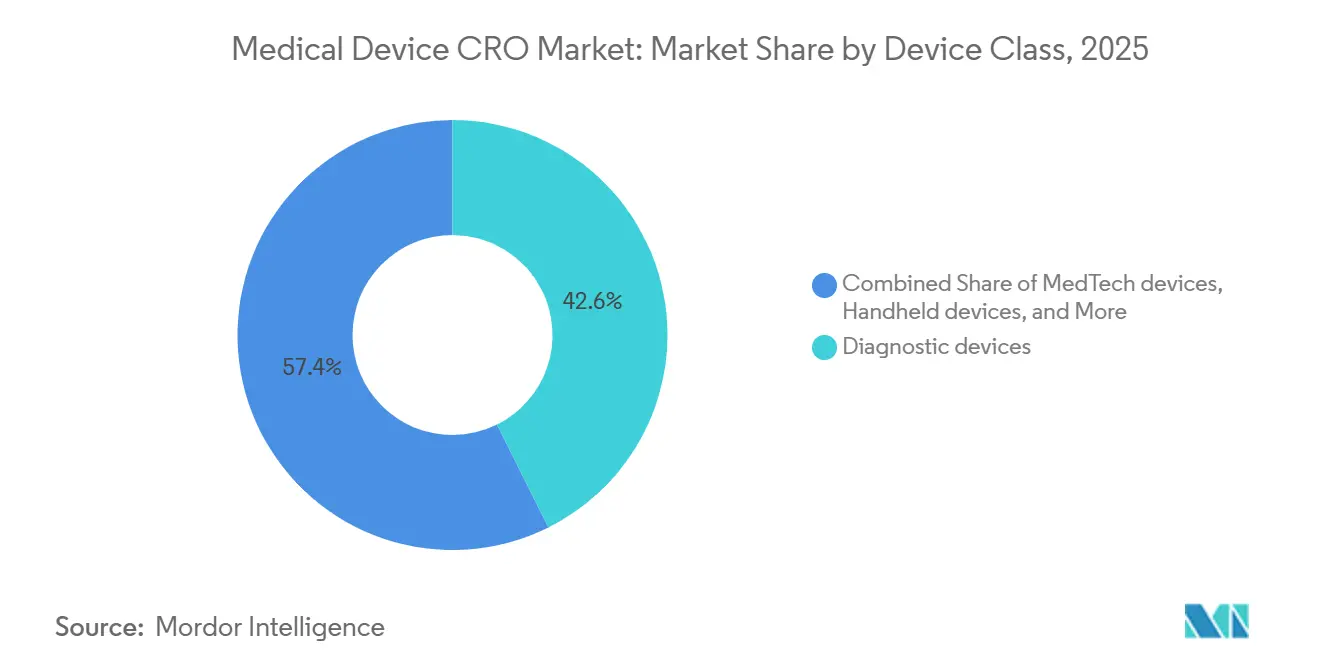
- Por classe de dispositivo, os dispositivos de diagnóstico representaram uma participação de 42,64% em 2025, enquanto os dispositivos MedTech estão projetados para registrar o maior CAGR de 7,42% até 2031.
- Por geografia, a América do Norte detinha 45,34% do mercado em 2025, enquanto a Ásia-Pacífico deve registrar o CAGR regional mais rápido de 8,32% até 2031.
Nota: O tamanho do mercado e os números de previsão neste relatório são gerados usando a estrutura de estimativa proprietária da Mordor Intelligence, atualizada com os dados e percepções mais recentes disponíveis em janeiro de 2026.
Tendências e Perspectivas do Mercado Global de CRO de Dispositivos Médicos
Análise de Impacto dos Impulsionadores*
| Impulsionador | (~) % de Impacto na Previsão de CAGR | Relevância Geográfica | Prazo de Impacto |
|---|---|---|---|
| A Terceirização se Intensifica à Medida que os Ensaios de Dispositivos Crescem em Volume e Complexidade | +2.0% | Global, mais agudo na América do Norte e na Europa Ocidental | Curto prazo (≤ 2 anos) |
| Os Requisitos de Evidências do MDR/IVDR da UE e as Exigências de PMCF Ampliam a Demanda | +1.8% | Europa como mercado primário, com repercussão na Ásia-Pacífico e na América do Norte para registros globais | Médio prazo (2 a 4 anos) |
| Os Requisitos de Cibersegurança da FDA e o Alinhamento com o QMSR Elevam as Necessidades de Validação de Software | +0.9% | América do Norte como mercado primário, com acompanhamento da UE e da Ásia-Pacífico | Médio prazo (2 a 4 anos) |
| A Adoção de Ensaios Descentralizados em Dispositivos Aumenta a Terceirização de Tecnologias de Saúde Digital e eClinical | +1.2% | América do Norte e Europa como mercados centrais, adoção inicial na Ásia-Pacífico | Curto prazo (≤ 2 anos) a Médio prazo (2 a 4 anos) |
| As Restrições de Transferência de Dados Transfronteiriços Impulsionam os Serviços de Engenharia de Privacidade | +0.7% | UE, com relevância para programas transfronteiriços da América do Norte para a UE | Longo prazo (≥ 4 anos) |
| Fonte: Mordor Intelligence | |||
A Terceirização se Intensifica à Medida que os Ensaios de Dispositivos Crescem em Volume e Complexidade
O mercado de CRO de dispositivos médicos está se beneficiando de um modelo de evidências clínicas que agora se estende muito além da submissão pré-mercado, porque a vigilância pós-mercado e o PMCF são obrigações contínuas e não atividades pontuais. O MDCG 2025-10 estabelece que o sistema de vigilância pós-mercado deve coletar e analisar dados ativamente ao longo de toda a vida útil do dispositivo, e que essas informações devem retroalimentar os processos de gestão de riscos, avaliação clínica, documentação técnica e ações corretivas. Esse requisito torna mais difícil para os fabricantes manter todas as tarefas regulatórias, bioestatísticas, de monitoramento e de relatórios dentro de suas próprias equipes, especialmente quando produtos de maior risco também exigem relatórios estruturados e atualizações contínuas.
O mercado de CRO de dispositivos médicos, portanto, se beneficia de trabalhos recorrentes em produtos legados, bem como em novos lançamentos, uma vez que dispositivos mais antigos que migram para novos marcos regulatórios podem reabrir lacunas de evidências e desencadear novos mandatos de terceirização. A Medpace reportou receita total de 2025 de USD 2.530,2 milhões, um aumento de 20%, e orientou para USD 2,755 bilhões a USD 2,855 bilhões em 2026, o que corrobora a visão de que a demanda por terceirização permaneceu saudável em toda a base de serviços de desenvolvimento mais ampla[1]Grupo de Coordenação de Dispositivos Médicos, "Orientação MDCG 2025-10 sobre Vigilância Pós-Mercado de Dispositivos Médicos e Dispositivos Médicos para Diagnóstico In Vitro", Comissão Europeia, health.ec.europa.eu.
Requisitos de Evidências do MDR/IVDR da UE e as Exigências de PMCF Ampliam a Demanda
O MDCG 2025-10 torna o sistema de vigilância pós-mercado parte integrante do sistema de gestão da qualidade, o que significa que o trabalho de evidências clínicas agora está inserido em um ciclo de conformidade contínuo, em vez de ser um exercício de arquivamento independente. A orientação também estabelece que um plano de PMCF ou PMPF deve ser incluído no plano de vigilância pós-mercado, ou o fabricante deve justificar por que essa atividade não é aplicável. Os resultados do PMCF e do PMPF devem então atualizar a avaliação clínica ou de desempenho, a avaliação de benefício-risco, a rotulagem, a documentação técnica e outros registros de conformidade contínuos. No mercado de CRO de dispositivos médicos, essa estrutura aumenta a demanda por monitoramento, gestão de dados, redação médica, suporte à vigilância e manutenção regulatória, porque os patrocinadores devem manter as evidências atualizadas após o lançamento e não apenas antes da aprovação. Os fornecedores que conseguem combinar capacidades de evidências do mundo real com fluxos de trabalho de documentação estão melhor posicionados à medida que os patrocinadores transferem mais desse trabalho para fora das equipes internas que já estão sobrecarregadas pelas obrigações do MDR e do IVDR.
Os Requisitos de Cibersegurança da FDA e o Alinhamento com o QMSR Elevam as Necessidades de Validação de Software e Testes de Cibersegurança
A orientação de cibersegurança da FDA de fevereiro de 2026 fornece recomendações sobre design, rotulagem e conteúdo de submissão para dispositivos com risco de cibersegurança, e aborda as expectativas da seção 524B para dispositivos cibernéticos. Esse documento substituiu a orientação de junho de 2025, o que sinaliza que as expectativas de cibersegurança estão evoluindo para um padrão operacional mais formal e atualizado para submissões de dispositivos. Ao mesmo tempo, o QMSR entrou em vigor em 2 de fevereiro de 2026 e incorporou a ISO 13485:2016, enfatizando a gestão de riscos ao longo dos processos de design, desenvolvimento, produção e outros processos do ciclo de vida. A regra do Registro Federal também adicionou requisitos detalhados de arquivos de reclamações, registros de manutenção e documentação vinculada ao UDI, que aumentam a carga de trabalho para programas de dispositivos conectados e ricos em software. Essa combinação está ampliando o papel de fornecedores especializados em validação de software e testes de cibersegurança no mercado de CRO de dispositivos médicos, porque muitos patrocinadores não dispõem das ferramentas ou do pessoal interno para manter esses fluxos de trabalho atualizados em escala[2]Administração de Alimentos e Medicamentos, "Dispositivos Médicos, Emendas ao Regulamento do Sistema de Qualidade", Registro Federal, govinfo.gov.
A Adoção de Ensaios Descentralizados em Dispositivos Aumenta a Terceirização de Tecnologias de Saúde Digital/eClinical e Operações Remotas
A adoção de ensaios descentralizados está se tornando cada vez mais relevante para o mercado de CRO de dispositivos médicos, porque os dispositivos representaram 21% dos registros de ensaios clínicos descentralizados intervencionais em uma revisão global de 1.370 estudos. A mesma análise mostrou que apenas uma pequena minoria dos ensaios clínicos descentralizados operava sem ferramentas digitais, o que confirma que a execução remota de ensaios agora depende de sistemas de dados, ferramentas de comunicação e fluxos de trabalho digitais. Ao mesmo tempo, a atividade de ensaios clínicos descentralizados permaneceu concentrada em países de alta renda, o que sugere que os modelos remotos estão se expandindo dentro de sistemas de pesquisa estabelecidos antes de ampliar totalmente o acesso a novos grupos de pacientes. Esse padrão ainda sustenta a demanda por terceirização, porque os patrocinadores precisam de ajuda com sistemas eClinical, coordenação remota de centros, tratamento distribuído de dados e métodos de monitoramento híbrido em várias geografias. O mercado de CRO de dispositivos médicos, portanto, se beneficia da adoção de ensaios clínicos descentralizados, mas os vencedores provavelmente serão os fornecedores que conseguem combinar operações de ensaios digitais com as necessidades práticas de manuseio que os estudos de dispositivos ainda exigem.
Análise de Impacto das Restrições*
| Restrição | (~) % de Impacto na Previsão de CAGR | Relevância Geográfica | Prazo de Impacto |
|---|---|---|---|
| Os Gargalos de Capacidade dos Organismos Notificados Prolongam as Certificações na UE | -0.8% | Europa como mercado central, impacto secundário nas submissões duplas da América do Norte para a UE | Curto prazo (≤ 2 anos) a Médio prazo (2 a 4 anos) |
| Os Obstáculos de Recrutamento de Pacientes, Acesso à Tecnologia e Governança de Dados nos Ensaios Descentralizados Retardam a Execução | -0.5% | Global, mais pronunciado em mercados emergentes de menor renda e da Ásia-Pacífico | Médio prazo (2 a 4 anos) |
| As Obrigações de SBOM de Cibersegurança e Aplicação de Patches Adicionam Ônus Recorrente de Revalidação | -0.4% | América do Norte como mercado primário, fator de conformidade emergente na UE | Longo prazo (≥ 4 anos) |
| Fonte: Mordor Intelligence | |||
Os Gargalos de Capacidade dos Organismos Notificados Prolongam as Certificações na UE
A capacidade dos organismos notificados continua sendo um dos riscos de cronograma mais evidentes para o mercado de CRO de dispositivos médicos na Europa, porque o fluxo de certificações ainda está aquém do volume de trabalho que passa pelos processos do MDR e do IVDR. A MedTech Europe descreveu os organismos notificados como um gargalo regulatório, e essa caracterização está alinhada com a pressão sustentada sobre os patrocinadores que precisam de aprovações, renovações e atualizações de evidências sob regras mais rígidas. Quando o cronograma de certificação atrasa, os contratos vinculados a marcos podem atrasar o reconhecimento de receita para as CROs, mesmo que o trabalho clínico ou de documentação já tenha sido concluído. Isso não elimina a demanda do mercado de CRO de dispositivos médicos, mas torna os cronogramas de entrega europeus mais difíceis de prever e aumenta o valor dos fornecedores que conseguem combinar trabalho clínico com coordenação regulatória estreita. Na prática, o gargalo desloca a concorrência para empresas que conseguem gerenciar o risco de acúmulo, sequenciar os fluxos de trabalho com cuidado e manter a confiança do patrocinador durante longos ciclos de revisão[3]MedTech Europe, "Os Organismos Notificados Estão se Tornando um Gargalo Regulatório", MedTech Europe, medtecheurope.org.
Os Obstáculos de Recrutamento de Pacientes, Acesso à Tecnologia e Governança de Dados nos Ensaios Descentralizados Retardam a Execução dos Estudos
Os modelos descentralizados podem ampliar o alcance dos ensaios, mas as evidências ainda mostram que a maior parte da atividade de ensaios clínicos descentralizados em um único país está concentrada em ambientes de alta renda, em vez de em grupos de pacientes carentes. Essa concentração significa que os benefícios da execução remota são desiguais, especialmente quando os patrocinadores esperam ganhos rápidos de recrutamento em mercados com acesso digital e capacidades de centros muito diferentes. A Comissão Europeia esclareceu que tanto o Regulamento de Ensaios Clínicos quanto o RGPD se aplicam simultaneamente, o que adiciona obrigações sobre base legal, armazenamento, arquivamento, direitos dos participantes, medidas de segurança e transferências internacionais de dados. O Comitê Europeu para a Proteção de Dados também tratou essa interação como uma questão de conformidade distinta, o que reforça como a governança de dados pode retardar o início dos estudos quando várias partes, sistemas e países estão envolvidos. Para o mercado de CRO de dispositivos médicos, isso significa que a execução de ensaios clínicos descentralizados ainda precisa de mais suporte jurídico, operacional e técnico do que muitos patrocinadores inicialmente presumiram, e isso reduz a vantagem de velocidade que os modelos remotos podem oferecer de outra forma.
*Nossas previsões tratam os impactos dos impulsionadores e restrições como direcionais, e não aditivos. As previsões de impacto refletem o crescimento de base, os efeitos de composição e as interações entre variáveis.
Análise de Segmentos
Por Escala de Operação: Os Serviços Clínicos Definem a Agenda de Terceirização
Os serviços clínicos detinham 58,46% da participação do mercado de CRO de dispositivos médicos em 2025, e o mesmo segmento está projetado para crescer a um CAGR de 7,67% até 2031. Essa posição de liderança reflete mais do que o simples volume de ensaios, porque os fabricantes agora enfrentam obrigações clínicas que continuam no período pós-mercado e retroalimentam a gestão de riscos e a documentação técnica. O MDCG 2025-10 torna essa continuidade explícita ao tratar a vigilância pós-mercado como um sistema ativo e contínuo que deve atualizar a avaliação de benefício-risco, a avaliação clínica e as ações corretivas ao longo de toda a vida útil do dispositivo. Para o mercado de CRO de dispositivos médicos, isso significa que o trabalho clínico não está mais concentrado apenas em torno da primeira submissão, uma vez que o PMCF e a manutenção de evidências relacionadas estendem a atividade muito após o lançamento. O resultado é um conjunto de terceirização maior e mais duradouro para patrocinadores com produtos Classe IIb, Classe III, implantáveis e ricos em software que não conseguem suportar essas tarefas internamente no ritmo exigido.
Os serviços pré-clínicos e de testes permanecem menores dentro do mercado de CRO de dispositivos médicos, mas estão se tornando mais estrategicamente importantes porque estão a montante de muitas decisões regulatórias e clínicas posteriores. Os patrocinadores de dispositivos ainda precisam de planos de avaliação biológica, caracterização química, avaliações toxicológicas e análises de lacunas que estejam alinhadas com as expectativas regulatórias atuais, em vez de suposições de aprovação mais antigas. A oferta declarada de biocompatibilidade da Labcorp mostra como esses serviços já cobrem testes de acordo com a ISO 10993, caracterização química, avaliação de risco toxicológico e suporte para revisões de lacunas de dispositivos legados vinculadas aos padrões regulatórios atuais. Isso mantém o setor de CRO de dispositivos médicos dependente de fornecedores de testes que conseguem conectar o trabalho de bancada à estratégia de submissão, em vez de atuar apenas como laboratórios isolados no início do desenvolvimento.


Por Tipo de Serviço: Assuntos Regulatórios e Médicos Superam Todas as Outras Linhas de Serviço
O Monitoramento Clínico manteve uma participação de 21,29% do tamanho do mercado de CRO de dispositivos médicos em 2025, enquanto Assuntos Regulatórios e Médicos está projetado para expandir a um CAGR de 7,82% até 2031. Esse padrão de crescimento reflete a profundidade das mudanças regulatórias atuais nos Estados Unidos e na Europa, onde os patrocinadores agora precisam de mais suporte contínuo em documentação, alinhamento de qualidade, submissões e manutenção pós-mercado. O QMSR entrou em vigor em 2026 e incorporou a ISO 13485:2016 na 21 CFR Parte 820, ao mesmo tempo em que preservou os controles de registros específicos da FDA, como arquivos de reclamações, registros de manutenção e documentação vinculada ao UDI. Esses requisitos tornam o trabalho regulatório mais contínuo, porque os patrocinadores devem manter sistemas e registros em conformidade ao longo de toda a vida útil do produto, em vez de montar um arquivo de submissão restrito em um único momento. No mercado de CRO de dispositivos médicos, isso transforma Assuntos Regulatórios e Médicos em um motor de crescimento para fornecedores que conseguem combinar qualidade, rotulagem, documentação e suporte voltado ao regulador em um único modelo operacional.
O Monitoramento Clínico ainda sustenta a receita no mercado de CRO de dispositivos médicos porque cada mudança regulatória mais ampla ainda depende de supervisão confiável de centros, fluxo de dados e escalada de problemas durante estudos ativos. O uso crescente de ferramentas digitais e modelos remotos está mudando a forma como o monitoramento é realizado, mas não reduziu a necessidade de controle operacional disciplinado em programas de dispositivos. Gestão de Dados e Bioestatística, Redação Médica e Recrutamento de Pacientes e Centros continuam a atuar como funções habilitadoras que tornam possível a entrega clínica e regulatória em escala. Dentro do setor de CRO de dispositivos médicos, os prestadores de serviços que integram essas funções de forma estreita estão em melhor posição do que as empresas que vendem cada fluxo de trabalho como uma tarefa separada com equipes e cronogramas distintos.
Por Classe de Dispositivo: Os Dispositivos de Diagnóstico Lideram, mas os Dispositivos MedTech Aceleram
Os dispositivos de diagnóstico representaram 42,64% do mercado em 2025, o que os manteve como a maior classe de dispositivos dentro do mercado de CRO de dispositivos médicos. Sua liderança está intimamente ligada à pressão do IVDR, porque uma parcela muito maior dos diagnósticos in vitro agora requer o envolvimento de organismos notificados do que sob a diretiva anterior. Essa mudança aumenta a quantidade de evidências de desempenho, trabalho de documentação e preparação do sistema de qualidade que os fabricantes devem concluir antes e após o acesso ao mercado. O mercado de CRO de dispositivos médicos, portanto, registra forte demanda de diagnósticos não apenas de novos produtos, mas também de produtos que passam por processos de reclassificação e transição que criam trabalho adicional de evidências. Essa é uma das razões pelas quais os diagnósticos continuam a gerar demanda estável de terceirização, mesmo quando o cronograma de certificação permanece irregular em toda a Europa.
Os dispositivos MedTech estão projetados para crescer mais rapidamente a um CAGR de 7,42% até 2031, o que reflete a crescente atividade em torno de sistemas conectados, produtos orientados por software e plataformas implantáveis complexas. As expectativas atuais da FDA em matéria de cibersegurança e sistema de gestão da qualidade aumentam a quantidade de validação, documentação e gestão de riscos ao longo do ciclo de vida exigida para esses produtos antes que os patrocinadores possam avançar de forma eficiente pela revisão e manutenção pós-mercado. Os produtos portáteis e de uso doméstico também estão adicionando trabalho no mercado de CRO de dispositivos médicos porque frequentemente precisam de pacotes de evidências que conectem as condições de uso no mundo real com as expectativas regulatórias e de qualidade. Dentro do setor de CRO de dispositivos médicos, o mix está, portanto, se deslocando para fornecedores que conseguem lidar com validação de software, segurança biológica, evidências clínicas e redação regulatória sem forçar os patrocinadores a coordenar vários especialistas desconectados.

Análise Geográfica
A América do Norte detinha 45,34% da participação do mercado de CRO de dispositivos médicos em 2025, o que a tornou o maior contribuinte regional. Os Estados Unidos respondem pela maior parte dessa posição porque o calendário regulatório comprimiu várias exigências de conformidade em um curto período, especialmente por meio da implementação do QMSR e das expectativas atualizadas de cibersegurança. O QMSR agora incorpora a ISO 13485:2016 por referência e também mantém os controles de registros específicos da FDA, o que significa que os fabricantes devem gerenciar a harmonização sem presumir que a certificação ISO por si só substitui a inspeção da FDA ou as obrigações de documentação. Muitos patrocinadores estão preenchendo essa lacuna por meio da terceirização, em vez de depender apenas da contratação interna de garantia da qualidade e assuntos regulatórios, o que ajuda a sustentar o mercado de CRO de dispositivos médicos da América do Norte mesmo em um ambiente de gastos mais disciplinado. Isso torna a região o exemplo mais claro de como a regulação pode expandir a demanda por terceirização por meio da complexidade de conformidade, e não apenas pelo volume de ensaios.
A Europa permanece o segundo maior cluster regional no mercado de CRO de dispositivos médicos, e é a geografia onde a regulação remodela mais diretamente o mix de serviços. O MDR e o IVDR impulsionaram mais evidências pós-mercado, manutenção de documentação e interação com organismos notificados para o modelo operacional, o que aumenta a dependência de CROs que conseguem combinar trabalho clínico e regulatório. Ao mesmo tempo, os gargalos dos organismos notificados continuam a complicar o cronograma, o que significa que a demanda por CRO permanece forte mesmo enquanto a conversão de contratos e o ritmo de marcos se tornam mais difíceis de prever. Isso deixa a Europa como uma região com força de demanda estrutural no mercado de CRO de dispositivos médicos, mas também com risco de cronograma de receita mais visível do que a América do Norte.
A Ásia-Pacífico está prevista para registrar o CAGR mais rápido de 8,32% no tamanho do mercado de CRO de dispositivos médicos até 2031. A região se beneficia da expansão da atividade de desenvolvimento de dispositivos, mas os processos regulatórios locais ainda tornam a capacidade local importante e não opcional. A PMDA mantém estruturas formais de notificação e consulta de ensaios clínicos para dispositivos médicos, e esses processos reforçam a necessidade de tratamento regulatório localizado, em vez de um único modelo global. Isso sustenta a demanda no mercado de CRO de dispositivos médicos por fornecedores que conseguem combinar acesso regional a centros, suporte linguístico e execução voltada ao regulador no Japão e nos mercados vizinhos. A China e a Índia também estão ampliando a base de demanda futura à medida que os pipelines de fabricação doméstica criam mais trabalho clínico e regulatório que os patrocinadores podem preferir terceirizar. A Coreia do Sul acrescenta a esse apelo regional porque um ambiente de aprovação mais eficiente pode suportar modelos de ensaios híbridos e footprints de estudos regionais mais amplos. O Oriente Médio e a África, juntamente com a América do Sul, permanecem menores no mercado de CRO de dispositivos médicos, mas os programas de registro multinacional nos países do CCG, no Brasil e na Argentina ainda criam espaço para fornecedores internacionais com alcance operacional local.

Cenário Competitivo
O mercado de CRO de dispositivos médicos é moderadamente fragmentado, com um número limitado de organizações globais de serviço completo e um campo muito mais amplo de empresas especializadas focadas em testes, assuntos regulatórios ou validação digital. Essa estrutura é importante porque os patrocinadores de dispositivos frequentemente precisam de várias disciplinas ao mesmo tempo, e nem todo fornecedor consegue conectar segurança biológica, operações clínicas, validação de software e suporte à submissão em um único contrato. A consolidação está, portanto, se tornando uma ferramenta competitiva prática no mercado de CRO de dispositivos médicos, especialmente para empresas que tentam passar de conjuntos de capacidades restritas para entrega de ponta a ponta. A aquisição pela NAMSA das operações de testes de dispositivos médicos nos Estados Unidos da WuXi AppTec em fevereiro de 2025 expandiu a capacidade laboratorial e a amplitude dos serviços, enquanto seu acordo de janeiro de 2026 pelo negócio de testes de dispositivos médicos de desenvolvimento inicial da Labcorp fortaleceu a profundidade dos testes em microbiologia, biocompatibilidade e áreas relacionadas. A NAMSA também afirma que mais de 70% dos estudos globais de biocompatibilidade são conduzidos por sua organização, o que mostra como alguns nichos críticos de conformidade já se tornaram concentrados, mesmo que o mercado de CRO de dispositivos médicos mais amplo permaneça mais disperso.
Outros grandes fornecedores também estão ampliando seu alcance por meio de movimentos de portfólio direcionados, em vez de depender apenas da expansão orgânica. A ICON divulgou sua aquisição de USD 92,5 milhões do Grupo KCR S.A. em 2024, e em dezembro de 2025 anunciou um acordo para adquirir a ClinicalRM para aprofundar sua posição em pesquisas patrocinadas pelo governo e trabalhos de doenças infecciosas que também podem suportar programas de dispositivos. A integração completa do Medidata Clinical Data Studio pela ICON em março de 2025 também aponta para um caminho de diferenciação baseado em tecnologia, uma vez que a plataforma reúne dados de fontes Medidata e não Medidata em um único fluxo de trabalho para gestão de dados, revisão e monitoramento central. Essas etapas mostram que o mercado de CRO de dispositivos médicos não está competindo apenas em número de funcionários ou alcance geográfico, porque a profundidade da plataforma e a integração de fluxos de trabalho estão se tornando parte da oferta comercial.
A tecnologia está se tornando uma fronteira competitiva mais visível no mercado de CRO de dispositivos médicos porque os patrocinadores querem cada vez mais um único parceiro que possa trabalhar em revisão de dados, monitoramento, registros de qualidade e documentação regulatória sem transferências repetidas. Ao mesmo tempo, os fornecedores especializados ainda são importantes porque o trabalho com dispositivos inclui categorias restritas, mas essenciais, como biocompatibilidade, extraíveis e lixiviáveis, e validação focada em cibersegurança, que as empresas generalistas maiores nem sempre controlam internamente. O mercado está, portanto, vendo dois caminhos ao mesmo tempo, com grandes empresas se expandindo por meio de aquisições e integração, enquanto empresas de nicho defendem sua posição por meio de profundidade técnica e resposta mais rápida em linhas de serviço direcionadas. O espaço em branco permanece mais visível onde dispositivos ricos em software requerem suporte alinhado em evidências clínicas, tratamento de dados com consciência de privacidade e manutenção de qualidade ao longo do ciclo de vida. Isso mantém o mercado de CRO de dispositivos médicos competitivo, ativo e apenas moderadamente concentrado, apesar do papel mais forte de alguns players de escala em certos nichos de testes.
Líderes do Setor de CRO de Dispositivos Médicos
IQVIA
NAMSA
Medpace
Charles River Laboratories
Avania
- *Isenção de responsabilidade: Principais participantes classificados em nenhuma ordem específica

Desenvolvimentos Recentes do Setor
- Fevereiro de 2026: O QMSR da FDA (21 CFR Parte 820, conforme alterado) entrou em vigor em 2 de fevereiro, incorporando a ISO 13485:2016 por referência, aposentando a Técnica de Inspeção do Sistema de Qualidade (QSIT) em favor de um processo de inspeção atualizado sob o Programa de Conformidade 7382.850. O QMSR introduz integração obrigatória de gestão de riscos ao longo do ciclo de vida do produto e novos requisitos de manutenção de registros vinculados ao UDI, ampliando a demanda por CRO para serviços de avaliação de lacunas do sistema de gestão da qualidade e revalidação.
- Janeiro de 2026: A Labcorp anunciou a venda de seu negócio de testes de dispositivos médicos de Desenvolvimento Inicial para a NAMSA. A transação expande a capacidade de testes da NAMSA em microbiologia, biocompatibilidade e extraíveis/lixiviáveis, reforçando sua posição como a CRO de testes pré-clínicos dominante no setor de dispositivos médicos.


Escopo do Relatório do Mercado Global de CRO de Dispositivos Médicos
De acordo com o escopo do relatório, uma Organização de Pesquisa Contratada (CRO) de dispositivos médicos é uma empresa especializada que fornece serviços terceirizados a fabricantes de dispositivos médicos. Esses serviços apoiam o desenvolvimento, os testes e a aprovação regulatória de dispositivos médicos. As CROs ajudam a agilizar os ensaios clínicos, as submissões regulatórias e as atividades de conformidade, permitindo que os fabricantes levem seus dispositivos ao mercado de forma mais eficiente.
O mercado de CRO de dispositivos médicos é segmentado por escala de operação em fase pré-clínica/testes e fase clínica. Por tipo de serviço, o mercado é categorizado em serviços de monitoramento clínico, serviços de gestão de ensaios clínicos, serviços de gestão de dados e bioestatística, serviços de assuntos regulatórios e médicos, serviços de redação médica, serviços de recrutamento de pacientes e centros, serviços de segurança e farmacovigilância e outros serviços. Por classe de dispositivo, a segmentação inclui dispositivos MedTech, dispositivos de diagnóstico, dispositivos portáteis e outros dispositivos. Por geografia, o mercado é dividido na região da América do Norte, região europeia, região da Ásia-Pacífico, região do Oriente Médio e África e região da América do Sul. O relatório de mercado também abrange os tamanhos de mercado estimados e as tendências para 17 países nas principais regiões globalmente. Para cada segmento, o tamanho do mercado e a previsão são fornecidos em termos de valor (USD).
| Pré-clínico/Testes |
| Clínico |
| Monitoramento Clínico |
| Gestão de Ensaios Clínicos |
| Gestão de Dados e Bioestatística |
| Assuntos Regulatórios e Médicos |
| Redação Médica |
| Recrutamento de Pacientes e Centros |
| Segurança e Farmacovigilância |
| Outros (Habilitação de Ensaios Descentralizados e Plataformas eClinical, Imagem/Laboratório Central, etc.) |
| Dispositivos MedTech |
| Dispositivos de diagnóstico |
| Dispositivos portáteis |
| Outros |
| América do Norte | Estados Unidos |
| Canadá | |
| México | |
| Europa | Alemanha |
| Reino Unido | |
| França | |
| Itália | |
| Espanha | |
| Restante da Europa | |
| Ásia-Pacífico | China |
| Índia | |
| Japão | |
| Coreia do Sul | |
| Austrália | |
| Restante da Ásia-Pacífico | |
| Oriente Médio e África | CCG |
| África do Sul | |
| Restante do Oriente Médio e África | |
| América do Sul | Brasil |
| Argentina | |
| Restante da América do Sul |
| Por Escala de Operação | Pré-clínico/Testes | |
| Clínico | ||
| Por Tipo de Serviço | Monitoramento Clínico | |
| Gestão de Ensaios Clínicos | ||
| Gestão de Dados e Bioestatística | ||
| Assuntos Regulatórios e Médicos | ||
| Redação Médica | ||
| Recrutamento de Pacientes e Centros | ||
| Segurança e Farmacovigilância | ||
| Outros (Habilitação de Ensaios Descentralizados e Plataformas eClinical, Imagem/Laboratório Central, etc.) | ||
| Por Classe de Dispositivo | Dispositivos MedTech | |
| Dispositivos de diagnóstico | ||
| Dispositivos portáteis | ||
| Outros | ||
| Por Geografia | América do Norte | Estados Unidos |
| Canadá | ||
| México | ||
| Europa | Alemanha | |
| Reino Unido | ||
| França | ||
| Itália | ||
| Espanha | ||
| Restante da Europa | ||
| Ásia-Pacífico | China | |
| Índia | ||
| Japão | ||
| Coreia do Sul | ||
| Austrália | ||
| Restante da Ásia-Pacífico | ||
| Oriente Médio e África | CCG | |
| África do Sul | ||
| Restante do Oriente Médio e África | ||
| América do Sul | Brasil | |
| Argentina | ||
| Restante da América do Sul | ||


Principais Perguntas Respondidas no Relatório
O que está impulsionando o crescimento nos serviços de CRO de dispositivos médicos até 2031?
O crescimento está sendo sustentado pelo aumento da carga regulatória, por necessidades mais longas de evidências pós-mercado e pela evolução de USD 11,91 bilhões em 2026 para USD 16,82 bilhões até 2031 a um CAGR de 7,14%.
Qual área de serviço lidera a demanda terceirizada para patrocinadores de dispositivos?
Os serviços clínicos lideram com uma participação de 58,46% em 2025, e também são o segmento de escala de operação de crescimento mais rápido, com um CAGR de 7,67% até 2031.
Por que os assuntos regulatórios e médicos estão crescendo mais rapidamente do que outras linhas de serviço?
O QMSR, o MDR e o IVDR aumentaram a quantidade de documentação, alinhamento de qualidade e trabalho de conformidade ao longo do ciclo de vida, razão pela qual Assuntos Regulatórios e Médicos está projetado para crescer a 7,82% até 2031.
Qual classe de dispositivo cria a maior oportunidade de terceirização hoje?
Os dispositivos de diagnóstico detinham a maior participação de 42,64% em 2025 porque o IVDR expandiu as necessidades de evidências e documentação para uma parcela maior da categoria.
Qual região está se expandindo mais rapidamente para o trabalho terceirizado de desenvolvimento de dispositivos?
A Ásia-Pacífico está prevista para crescer mais rapidamente a 8,32% até 2031, sustentada pela crescente atividade de desenvolvimento regional e pela necessidade de execução regulatória localizada.
Quão concentrada é a concorrência entre os fornecedores de CRO de dispositivos médicos?
A concorrência é moderada, e não altamente concentrada, porque as empresas globais de serviço completo estão ativas, mas muitos fornecedores especializados ainda ocupam posições importantes em testes, trabalho regulatório e validação digital.
Página atualizada pela última vez em: