Tamaño y Participación del Mercado de CRO de Dispositivos Médicos
Visión General del Mercado
| Período de Estudio | 2020 - 2031 |
|---|---|
| Tamaño del Mercado (2026) | 11.91 Mil millones de dólares |
| Tamaño del Mercado (2031) | 16.82 Mil millones de dólares |
| Tasa de crecimiento (2026 - 2031) | 7.14% CAGR |
| Mercado de Crecimiento Más Rápido | Asia-Pacífico |
| Mercado Más Grande | América del Norte |
| Concentración del Mercado | Medio |
Jugadores principales *Nota aclaratoria: los principales jugadores no se ordenaron de un modo en especial Imagen © Mordor Intelligence. El uso requiere atribución según CC BY 4.0. | |
Análisis del Mercado de CRO de Dispositivos Médicos por Mordor Intelligence
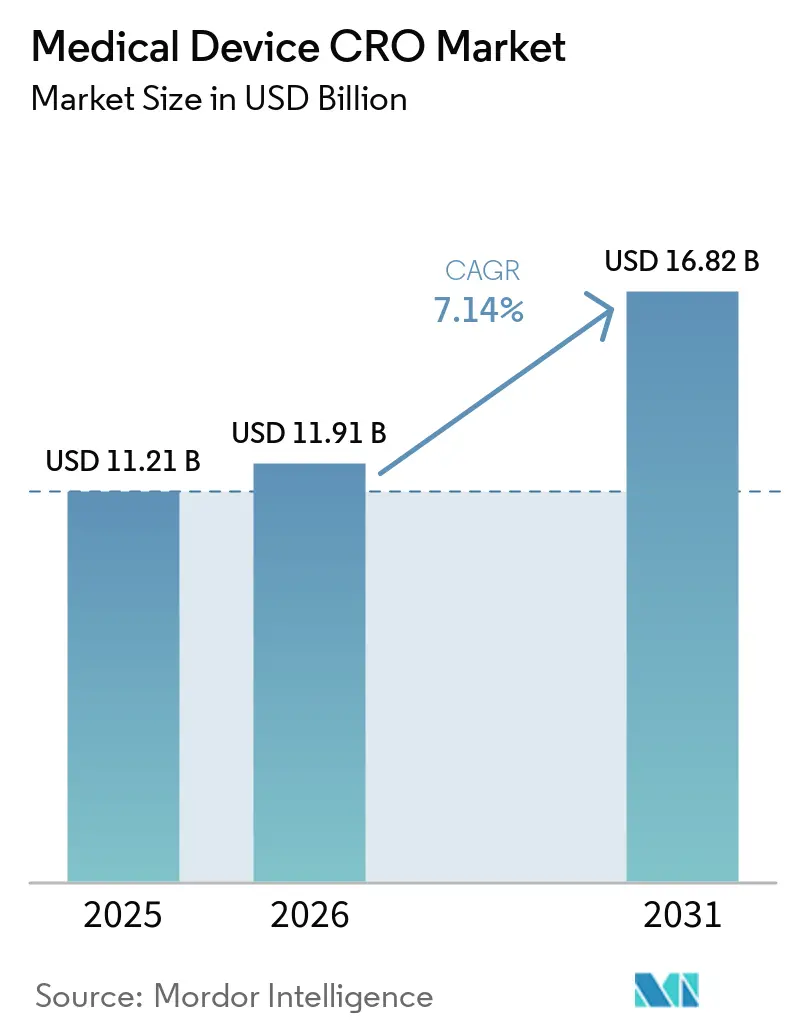
Se espera que el tamaño del Mercado de CRO de Dispositivos Médicos aumente de USD 11,21 mil millones en 2025 a USD 11,91 mil millones en 2026 y alcance USD 16,82 mil millones en 2031, creciendo a una CAGR del 7,14% durante 2026-2031.
El mercado de CRO de dispositivos médicos difiere de la externalización farmacéutica porque los patrocinadores de dispositivos frecuentemente necesitan validación de hardware, verificación de software, trabajo de usabilidad y pruebas de biocompatibilidad dentro del mismo paquete de presentación, lo que hace que las necesidades de externalización sean más fragmentadas y más exigentes desde el punto de vista técnico. El cambio al QMSR de la FDA en febrero de 2026 y la carga continua del MDR e IVDR de la UE están impulsando a los patrocinadores hacia socios especializados que puedan conectar la ingeniería, la ejecución clínica y el trabajo regulatorio dentro de un mismo programa, en lugar de en entregas separadas. El mercado de CRO de dispositivos médicos también permanece activo durante períodos más prolongados porque el desarrollo de dispositivos es iterativo, y las actualizaciones de diseño a menudo desencadenan nuevas rondas de documentación, pruebas y revalidación, en lugar de seguir un modelo limpio por fases. Las exigencias de gobernanza de datos transfronterizos y los cuellos de botella en la certificación en Europa todavía ralentizan los plazos para los flujos de trabajo multinacionales, especialmente cuando los patrocinadores necesitan paquetes de evidencia alineados entre regiones. La presión competitiva se mantiene de moderada a alta en el mercado de CRO de dispositivos médicos, y la reciente consolidación por parte de los proveedores de pruebas y servicios más grandes está reduciendo el espacio para las empresas de tamaño mediano que no pueden ofrecer una entrega integrada a lo largo del ciclo de vida.
Conclusiones Clave del Informe
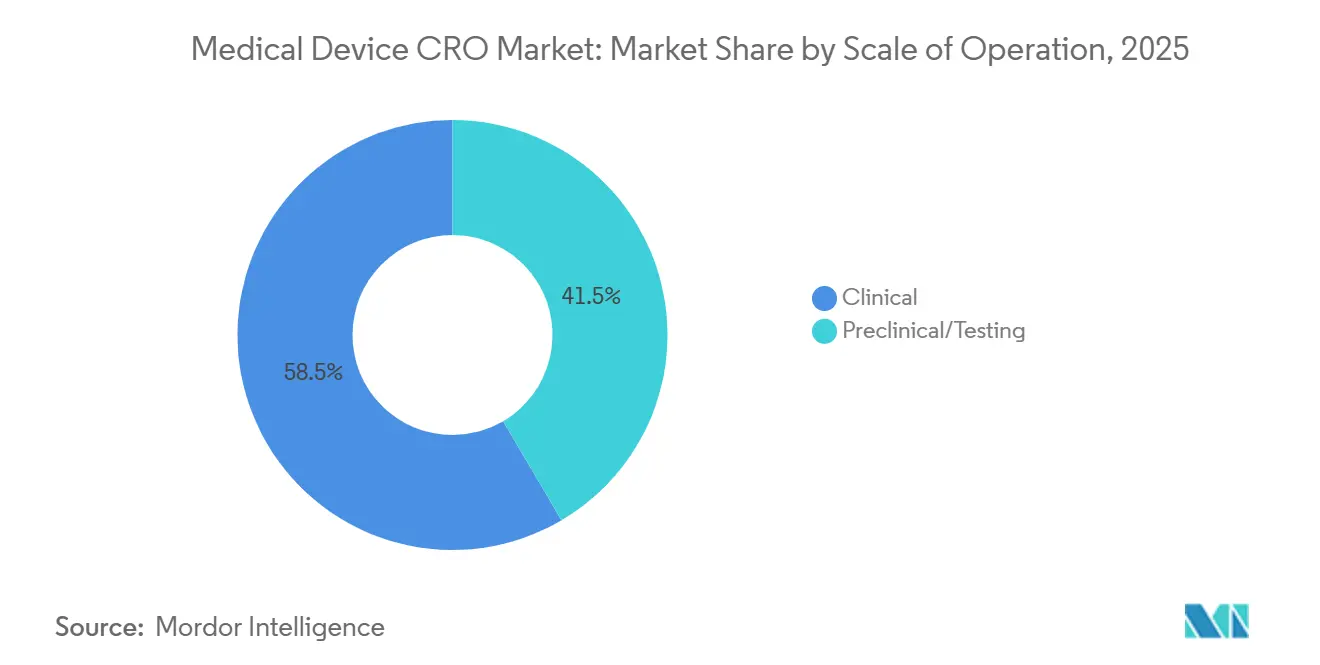
- Por escala de operación, los servicios clínicos representaron el 58,46% del mercado en 2025, y se proyecta que el mismo subsegmento se expanda a una CAGR del 7,67% hasta 2031.
- Por tipo de servicio, el Monitoreo Clínico lideró con una participación del 21,29% en 2025, mientras que se prevé que los Asuntos Regulatorios y Médicos crezcan más rápido con una CAGR del 7,82% hasta 2031.
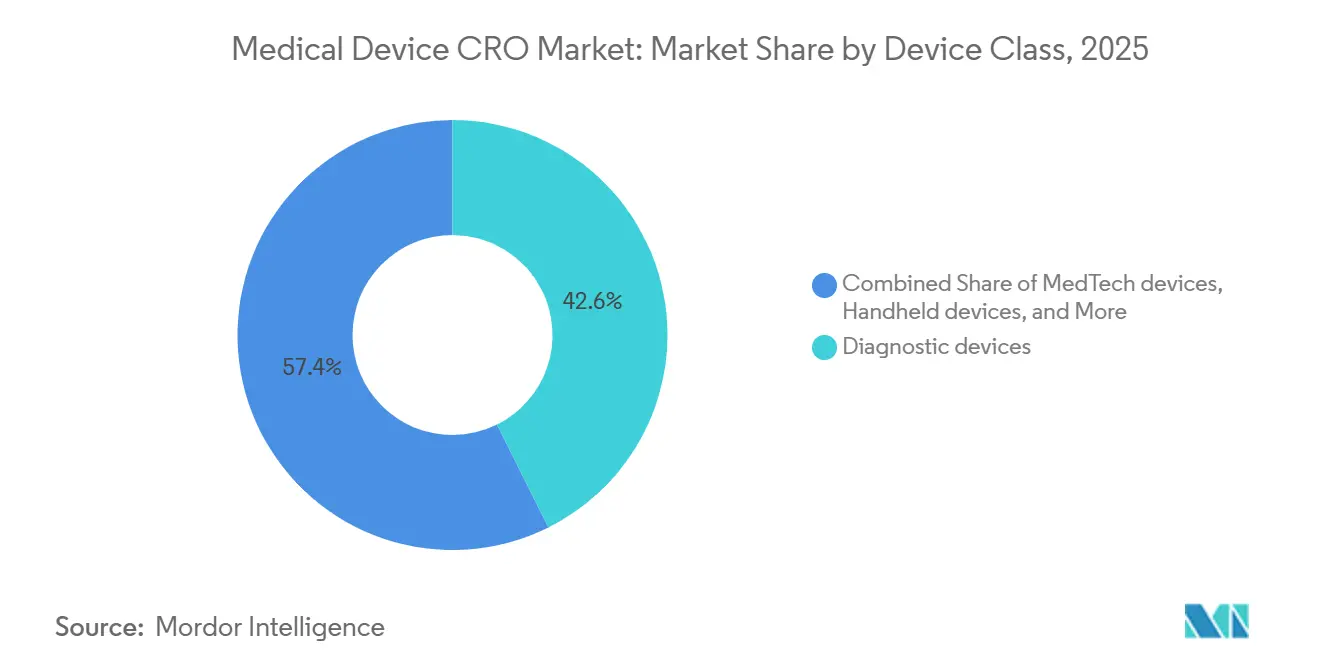
- Por clase de dispositivo, los dispositivos de diagnóstico representaron una participación del 42,64% en 2025, mientras que se proyecta que los dispositivos MedTech registren la CAGR más alta del 7,42% hasta 2031.
- Por geografía, América del Norte mantuvo el 45,34% del mercado en 2025, mientras que se espera que Asia-Pacífico registre la CAGR regional más rápida del 8,32% hasta 2031.
Nota: Las cifras del tamaño del mercado y los pronósticos de este informe se generan utilizando el marco de estimación patentado de Mordor Intelligence, actualizado con los datos y conocimientos más recientes disponibles a partir de enero de 2026.
Tendencias e Información del Mercado Global de CRO de Dispositivos Médicos
Análisis del Impacto de los Impulsores*
| Impulsor | (~) % de Impacto en el Pronóstico de CAGR | Relevancia Geográfica | Horizonte Temporal del Impacto |
|---|---|---|---|
| La Externalización se Intensifica a Medida que los Ensayos de Dispositivos Crecen en Volumen y Complejidad | +2.0% | Global, más agudo en América del Norte y Europa Occidental | Corto plazo (≤ 2 años) |
| Los Requisitos de Evidencia del MDR/IVDR de la UE y los Requisitos de PMCF Amplían la Demanda | +1.8% | Europa como mercado principal, con efecto secundario en APAC y América del Norte para registros globales | Mediano plazo (2-4 años) |
| Los Requisitos de Ciberseguridad de la FDA y la Alineación con el QMSR Elevan las Necesidades de Validación de Software | +0.9% | América del Norte como mercado principal, con seguimiento de la UE y APAC | Mediano plazo (2-4 años) |
| La Adopción de Ensayos Descentralizados en Dispositivos Aumenta la Externalización de DHT y eClinical | +1.2% | América del Norte y Europa como mercados principales, adopción temprana en APAC | Corto plazo (≤ 2 años) a Mediano plazo (2-4 años) |
| Las Restricciones de Transferencia de Datos Transfronterizos Impulsan los Servicios de Ingeniería de Privacidad | +0.7% | UE, con relevancia para los programas transfronterizos de América del Norte a la UE | Largo plazo (≥ 4 años) |
| Fuente: Mordor Intelligence | |||
La Externalización se Intensifica a Medida que los Ensayos de Dispositivos Crecen en Volumen y Complejidad
El mercado de CRO de dispositivos médicos se está beneficiando de un modelo de evidencia clínica que ahora se extiende mucho más allá de la presentación previa a la comercialización, porque la vigilancia poscomercialización y el PMCF son obligaciones continuas en lugar de actividades puntuales. El MDCG 2025-10 establece que el PMS debe recopilar y analizar activamente datos a lo largo de la vida útil del dispositivo, y que esta información debe retroalimentar los procesos de gestión de riesgos, evaluación clínica, documentación técnica y acciones correctivas. Ese requisito dificulta que los fabricantes mantengan todas las tareas regulatorias, bioestadísticas, de monitoreo e informes dentro de sus propios equipos, especialmente cuando los productos de mayor riesgo también requieren informes estructurados y actualizaciones continuas.
El mercado de CRO de dispositivos médicos se beneficia, por tanto, del trabajo recurrente sobre productos heredados, así como de nuevos lanzamientos, ya que los dispositivos más antiguos que se incorporan a marcos más nuevos pueden reabrir brechas de evidencia y desencadenar nuevos mandatos de externalización. Medpace reportó ingresos anuales completos de 2025 de USD 2.530,2 millones, un aumento del 20%, y orientó hacia USD 2.755 mil millones a USD 2.855 mil millones en 2026, lo que respalda la opinión de que la demanda de externalización se mantuvo saludable en toda la base de servicios de desarrollo más amplia[1]Grupo de Coordinación de Dispositivos Médicos, "Guía MDCG 2025-10 sobre Vigilancia Poscomercialización de Dispositivos Médicos y Dispositivos Médicos de Diagnóstico In Vitro," Comisión Europea, health.ec.europa.eu.
Evidencia del MDR/IVDR de la UE y los Requisitos de PMCF Amplían la Demanda
El MDCG 2025-10 convierte el sistema PMS en una parte integral del sistema de gestión de calidad, lo que significa que el trabajo de evidencia clínica ahora se sitúa dentro de un ciclo de cumplimiento continuo en lugar de ser un ejercicio de presentación independiente. La guía también establece que un plan de PMCF o PMPF debe incluirse dentro del plan PMS, o el fabricante debe justificar por qué esa actividad no es aplicable. Los hallazgos del PMCF y el PMPF deben actualizar la evaluación clínica o de desempeño, la evaluación de beneficio-riesgo, el etiquetado, la documentación técnica y otros registros de cumplimiento continuo. En el mercado de CRO de dispositivos médicos, esa estructura aumenta la demanda de monitoreo, gestión de datos, redacción médica, soporte de vigilancia y mantenimiento regulatorio, porque los patrocinadores deben mantener la evidencia actualizada después del lanzamiento y no solo antes de la aprobación. Los proveedores que pueden combinar capacidades de evidencia del mundo real con flujos de trabajo de documentación están mejor posicionados a medida que los patrocinadores transfieren más de este trabajo fuera de los equipos internos que ya están sobrecargados por las obligaciones del MDR e IVDR.
Los Requisitos de Ciberseguridad de la FDA y la Alineación con el QMSR Elevan las Necesidades de Validación de Software y Pruebas de Ciberseguridad
La guía de ciberseguridad de la FDA de febrero de 2026 ofrece recomendaciones sobre diseño, etiquetado y contenido de presentaciones para dispositivos con riesgo de ciberseguridad, y aborda las expectativas de la sección 524B para dispositivos cibernéticos. Ese documento reemplazó la guía de junio de 2025, lo que indica que las expectativas de ciberseguridad están evolucionando hacia un estándar operativo más formal y actualizado para las presentaciones de dispositivos. Al mismo tiempo, el QMSR entró en vigor el 2 de febrero de 2026 e incorporó la norma ISO 13485:2016, haciendo énfasis en la gestión de riesgos a través del diseño, el desarrollo, la producción y otros procesos del ciclo de vida. La norma del Registro Federal también añadió requisitos detallados de archivos de reclamaciones, registros de servicio y documentación vinculada al UDI que aumentan la carga de trabajo para los programas de dispositivos conectados y con gran contenido de software. Esta combinación está ampliando el papel de los proveedores especializados en validación de software y pruebas de ciberseguridad en el mercado de CRO de dispositivos médicos, porque muchos patrocinadores no cuentan con las herramientas internas ni el personal para mantener estos flujos de trabajo actualizados a escala[2]Administración de Alimentos y Medicamentos, "Dispositivos Médicos, Enmiendas al Reglamento del Sistema de Calidad," Registro Federal, govinfo.gov.
La Adopción de Ensayos Descentralizados en Dispositivos Aumenta la Externalización de DHT/eClinical y Operaciones Remotas
La adopción de ensayos descentralizados se está volviendo más relevante para el mercado de CRO de dispositivos médicos porque los dispositivos representaron el 21% de los registros de ensayos clínicos descentralizados intervencionistas en una revisión global de 1.370 estudios. El mismo análisis mostró que solo una pequeña minoría de los ensayos clínicos descentralizados operaba sin herramientas digitales, lo que confirma que la ejecución remota de ensayos ahora depende de sistemas de datos, herramientas de comunicación y flujos de trabajo digitales. Al mismo tiempo, la actividad de los ensayos clínicos descentralizados se mantuvo concentrada en países de ingresos altos, lo que sugiere que los modelos remotos se están expandiendo dentro de los sistemas de investigación establecidos antes de ampliar plenamente el acceso a nuevos grupos de pacientes. Ese patrón sigue respaldando la demanda de externalización, porque los patrocinadores necesitan ayuda con los sistemas eClinical, la coordinación remota de sitios, el manejo distribuido de datos y los métodos de monitoreo híbrido en varias geografías. El mercado de CRO de dispositivos médicos se beneficia, por tanto, de la adopción de ensayos clínicos descentralizados, pero es probable que los ganadores sean los proveedores que puedan combinar las operaciones de ensayos digitales con las necesidades prácticas de manejo que los estudios de dispositivos aún requieren.
Análisis del Impacto de las Restricciones*
| Restricción | (~) % de Impacto en el Pronóstico de CAGR | Relevancia Geográfica | Horizonte Temporal del Impacto |
|---|---|---|---|
| Los Cuellos de Botella en la Capacidad de los Organismos Notificados Prolongan las Certificaciones en la UE | -0.8% | Europa como mercado principal, impacto secundario en las presentaciones duales de América del Norte a la UE | Corto plazo (≤ 2 años) a Mediano plazo (2-4 años) |
| Los Obstáculos en el Reclutamiento de Pacientes, el Acceso Tecnológico y la Gobernanza de Datos en los Ensayos Clínicos Descentralizados Ralentizan la Ejecución | -0.5% | Global, más pronunciado en mercados emergentes de menores ingresos y APAC | Mediano plazo (2-4 años) |
| Las Obligaciones de SBOM de Ciberseguridad y Aplicación de Parches Añaden una Carga Recurrente de Revalidación | -0.4% | América del Norte como mercado principal, factor de cumplimiento emergente en la UE | Largo plazo (≥ 4 años) |
| Fuente: Mordor Intelligence | |||
Los Cuellos de Botella en la Capacidad de los Organismos Notificados Prolongan las Certificaciones en la UE
La capacidad de los organismos notificados sigue siendo uno de los riesgos de tiempo más evidentes para el mercado de CRO de dispositivos médicos en Europa, porque el flujo de certificaciones todavía está rezagado respecto al volumen de trabajo que se procesa a través de las vías del MDR e IVDR. MedTech Europe ha descrito a los organismos notificados como un cuello de botella regulatorio, y ese enfoque se alinea con la presión sostenida sobre los patrocinadores que necesitan aprobaciones, renovaciones y actualizaciones de evidencia bajo normas más estrictas. Cuando los plazos de certificación se retrasan, los contratos vinculados a hitos pueden retrasar el reconocimiento de ingresos para las CRO, incluso si el trabajo clínico o de documentación ya se ha completado. Esto no elimina la demanda del mercado de CRO de dispositivos médicos, pero sí hace que los calendarios de entrega europeos sean más difíciles de predecir y aumenta el valor de los proveedores que pueden combinar el trabajo clínico con una estrecha coordinación regulatoria. En términos prácticos, el cuello de botella desplaza la competencia hacia las empresas que pueden gestionar el riesgo de acumulación de trabajo, secuenciar los flujos de trabajo con cuidado y mantener la confianza del patrocinador durante largos ciclos de revisión[3]MedTech Europe, "Los Organismos Notificados se Están Convirtiendo en un Cuello de Botella Regulatorio," MedTech Europe, medtecheurope.org.
Los Obstáculos en el Reclutamiento de Pacientes, el Acceso Tecnológico y la Gobernanza de Datos en los Ensayos Clínicos Descentralizados Ralentizan la Ejecución de los Estudios
Los modelos descentralizados pueden ampliar el alcance de los ensayos, pero la evidencia sigue mostrando que la mayor parte de la actividad de ensayos clínicos descentralizados en un solo país está concentrada en entornos de ingresos altos en lugar de en grupos de pacientes desatendidos. Esa concentración significa que los beneficios de la ejecución remota son desiguales, especialmente cuando los patrocinadores esperan ganancias rápidas en la inscripción en mercados con acceso digital y capacidades de sitio muy diferentes. La Comisión Europea ha aclarado que tanto el Reglamento de Ensayos Clínicos como el RGPD se aplican al mismo tiempo, lo que añade obligaciones sobre la base legal, el almacenamiento, el archivo, los derechos de los sujetos, las medidas de seguridad y las transferencias internacionales de datos. El Comité Europeo de Protección de Datos también trató esta interacción como un problema de cumplimiento distinto, lo que refuerza cómo la gobernanza de datos puede ralentizar el inicio de los estudios cuando intervienen varias partes, sistemas y países. Para el mercado de CRO de dispositivos médicos, eso significa que la ejecución de ensayos clínicos descentralizados todavía necesita más apoyo legal, operativo y técnico del que muchos patrocinadores asumieron inicialmente, y esto reduce la ventaja de velocidad que los modelos remotos pueden ofrecer de otro modo.
*Nuestras previsiones consideran los impactos de impulsores y restricciones como direccionales, no aditivos. Las previsiones de impacto reflejan el crecimiento base, los efectos de mezcla y las interacciones entre variables.
Análisis de Segmentos
Por Escala de Operación: Los Servicios Clínicos Definen la Agenda de Externalización
Los servicios clínicos representaron el 58,46% de la participación del mercado de CRO de dispositivos médicos en 2025, y se proyecta que el mismo segmento crezca a una CAGR del 7,67% hasta 2031. Esta posición de liderazgo refleja más que el simple volumen de ensayos, porque los fabricantes ahora enfrentan obligaciones clínicas que continúan en el período poscomercialización y se retroalimentan en la gestión de riesgos y la documentación técnica. El MDCG 2025-10 hace explícita esa continuidad al tratar el PMS como un sistema activo y continuo que debe actualizar la evaluación de beneficio-riesgo, la evaluación clínica y las acciones correctivas a lo largo de la vida útil del dispositivo. Para el mercado de CRO de dispositivos médicos, eso significa que el trabajo clínico ya no se concentra únicamente en torno a la primera presentación, ya que el PMCF y el mantenimiento de evidencia relacionado extienden la actividad mucho después del lanzamiento. El resultado es un grupo de externalización más amplio y duradero para los patrocinadores con productos de Clase IIb, Clase III, implantables y con gran contenido de software que no pueden respaldar estas tareas internamente al ritmo requerido.
Los servicios preclínicos y de pruebas siguen siendo menores dentro del mercado de CRO de dispositivos médicos, aunque se están volviendo más estratégicamente importantes porque se sitúan en la parte inicial de muchas decisiones regulatorias y clínicas posteriores. Los patrocinadores de dispositivos todavía necesitan planes de evaluación biológica, caracterización química, evaluaciones toxicológicas y análisis de brechas que se alineen con las expectativas regulatorias actuales en lugar de con supuestos de aprobación más antiguos. La oferta declarada de biocompatibilidad de Labcorp muestra cómo estos servicios ya cubren las pruebas ISO 10993, la caracterización química, la evaluación del riesgo toxicológico y el soporte para revisiones de brechas de dispositivos heredados vinculadas a los estándares regulatorios actuales. Esto mantiene a la industria de CRO de dispositivos médicos dependiente de los proveedores de pruebas que pueden conectar el trabajo de laboratorio con la estrategia de presentación, en lugar de actuar únicamente como laboratorios aislados al inicio del desarrollo.


Por Tipo de Servicio: Los Asuntos Regulatorios y Médicos Superan a Todas las Demás Líneas de Servicio
El Monitoreo Clínico mantuvo una participación del 21,29% del tamaño del mercado de CRO de dispositivos médicos en 2025, mientras que se proyecta que los Asuntos Regulatorios y Médicos se expandan a una CAGR del 7,82% hasta 2031. Ese patrón de crecimiento refleja la profundidad del cambio regulatorio actual en los Estados Unidos y Europa, donde los patrocinadores ahora necesitan más apoyo continuo en documentación, alineación de calidad, presentaciones y mantenimiento poscomercialización. El QMSR entró en vigor en 2026 e incorporó la norma ISO 13485:2016 en el 21 CFR Parte 820, al tiempo que preservó los controles de registros específicos de la FDA, como los archivos de reclamaciones, los registros de servicio y la documentación vinculada al UDI. Estos requisitos hacen que el trabajo regulatorio sea más continuo, porque los patrocinadores deben mantener sistemas y registros conformes a lo largo de la vida del producto en lugar de ensamblar un archivo de presentación limitado en un momento determinado. En el mercado de CRO de dispositivos médicos, eso convierte a los Asuntos Regulatorios y Médicos en un motor de crecimiento para los proveedores que pueden combinar calidad, etiquetado, documentación y soporte orientado al regulador en un modelo operativo.
El Monitoreo Clínico sigue siendo el pilar de los ingresos en el mercado de CRO de dispositivos médicos porque cada cambio regulatorio más amplio sigue dependiendo de una supervisión confiable del sitio, el flujo de datos y la escalada de problemas durante los estudios activos. El creciente uso de herramientas digitales y modelos remotos está cambiando la forma en que se realiza el monitoreo, aunque no ha reducido la necesidad de un control operativo disciplinado en los programas de dispositivos. La Gestión de Datos y Bioestadística, la Redacción Médica y el Reclutamiento de Pacientes y Sitios continúan actuando como funciones habilitadoras que hacen posible la entrega clínica y regulatoria a escala. Dentro de la industria de CRO de dispositivos médicos, los proveedores de servicios que integran estas funciones de manera estrecha están en una mejor posición que las empresas que venden cada flujo de trabajo como una tarea separada con equipos y plazos independientes.
Por Clase de Dispositivo: Los Dispositivos de Diagnóstico Lideran, pero los Dispositivos MedTech se Aceleran
Los dispositivos de diagnóstico representaron el 42,64% del mercado en 2025, lo que los mantuvo como la clase de dispositivo más grande dentro del mercado de CRO de dispositivos médicos. Su liderazgo está estrechamente vinculado a la presión del IVDR, porque una proporción mucho mayor de los diagnósticos in vitro ahora requiere la participación de organismos notificados que bajo la directiva anterior. Ese cambio aumenta la cantidad de evidencia de desempeño, trabajo de documentación y preparación del sistema de calidad que los fabricantes deben completar antes y después del acceso al mercado. El mercado de CRO de dispositivos médicos, por tanto, registra una fuerte demanda de diagnósticos no solo de nuevos productos, sino también de productos que atraviesan vías de reclasificación y transición que crean trabajo de evidencia adicional. Esta es una de las razones por las que los diagnósticos continúan generando una demanda de externalización constante incluso cuando los plazos de certificación siguen siendo irregulares en toda Europa.
Se proyecta que los dispositivos MedTech crezcan más rápido con una CAGR del 7,42% hasta 2031, lo que refleja una actividad creciente en torno a sistemas conectados, productos impulsados por software y plataformas implantables complejas. Las expectativas actuales de la FDA en materia de ciberseguridad y sistemas de gestión de calidad aumentan la cantidad de validación, documentación y gestión de riesgos del ciclo de vida requerida para estos productos antes de que los patrocinadores puedan avanzar eficientemente a través de la revisión y el mantenimiento poscomercialización. Los productos portátiles y de uso doméstico también están añadiendo trabajo en el mercado de CRO de dispositivos médicos porque a menudo necesitan paquetes de evidencia que conecten las condiciones de uso en el mundo real con las expectativas regulatorias y de calidad. Dentro de la industria de CRO de dispositivos médicos, la combinación está, por tanto, cambiando hacia proveedores que puedan manejar la validación de software, la seguridad biológica, la evidencia clínica y la redacción regulatoria sin obligar a los patrocinadores a coordinar varios especialistas desconectados.

Análisis Geográfico
América del Norte mantuvo el 45,34% de la participación del mercado de CRO de dispositivos médicos en 2025, lo que la convirtió en el mayor contribuyente regional. Los Estados Unidos representan la mayor parte de esta posición porque el calendario regulatorio comprimió varias exigencias de cumplimiento en un período corto, especialmente a través de la implementación del QMSR y las expectativas actualizadas de ciberseguridad. El QMSR ahora incorpora la norma ISO 13485:2016 por referencia, y también mantiene en vigor los controles de registros específicos de la FDA, lo que significa que los fabricantes deben gestionar la armonización sin asumir que la certificación ISO por sí sola reemplaza la inspección de la FDA o los deberes de documentación. Muchos patrocinadores están cubriendo esta brecha a través de la externalización en lugar de depender únicamente de la contratación interna de aseguramiento de calidad y asuntos regulatorios, lo que ayuda a sostener el mercado de CRO de dispositivos médicos de América del Norte incluso en un entorno de gasto más disciplinado. Esto convierte a la región en el ejemplo más claro de cómo la regulación puede expandir la demanda de externalización a través de la complejidad del cumplimiento en lugar de solo a través del volumen de ensayos.
Europa sigue siendo el segundo grupo regional más grande en el mercado de CRO de dispositivos médicos, y es la geografía donde la regulación remodela más directamente la combinación de servicios. El MDR e IVDR han incorporado más evidencia poscomercialización, mantenimiento de documentación e interacción con organismos notificados en el modelo operativo, lo que aumenta la dependencia de las CRO que pueden combinar el trabajo clínico y regulatorio. Al mismo tiempo, los cuellos de botella de los organismos notificados continúan complicando los plazos, lo que significa que la demanda de CRO se mantiene fuerte incluso mientras la conversión de contratos y el ritmo de los hitos se vuelven más difíciles de predecir. Esto deja a Europa como una región con una fortaleza estructural de demanda en el mercado de CRO de dispositivos médicos, pero también con un riesgo de tiempo de ingresos más visible que América del Norte.
Se prevé que Asia-Pacífico registre la CAGR más rápida del 8,32% en el tamaño del mercado de CRO de dispositivos médicos hasta 2031. La región se beneficia de la expansión de la actividad de desarrollo de dispositivos, pero las vías regulatorias locales todavía hacen que la capacidad sobre el terreno sea importante en lugar de opcional. La PMDA mantiene estructuras formales de notificación y consulta de ensayos clínicos para dispositivos médicos, y esos procesos refuerzan la necesidad de un manejo regulatorio localizado en lugar de una plantilla global única. Esto respalda la demanda en el mercado de CRO de dispositivos médicos de proveedores que puedan combinar acceso regional a sitios, soporte lingüístico y ejecución orientada al regulador en Japón y los mercados vecinos. China e India también están ampliando la base de demanda futura a medida que las cadenas de fabricación nacionales crean más trabajo clínico y regulatorio que los patrocinadores pueden preferir externalizar. Corea del Sur añade atractivo a esta región porque un entorno de aprobación más eficiente puede respaldar modelos de ensayos híbridos y huellas de estudio regional más amplias. Oriente Medio y África, junto con América del Sur, siguen siendo menores en el mercado de CRO de dispositivos médicos, aunque los programas de registro multinacional en los países del CCG, Brasil y Argentina todavía crean espacio para los proveedores internacionales con alcance operativo local.

Panorama Competitivo
El mercado de CRO de dispositivos médicos está moderadamente fragmentado, con un número limitado de organizaciones globales de servicio completo y un campo mucho más amplio de empresas especializadas centradas en pruebas, asuntos regulatorios o validación digital. Esa estructura importa porque los patrocinadores de dispositivos a menudo necesitan varias disciplinas a la vez, y no todos los proveedores pueden conectar la seguridad biológica, las operaciones clínicas, la validación de software y el soporte de presentaciones en un solo compromiso. La consolidación se está convirtiendo, por tanto, en una herramienta competitiva práctica en el mercado de CRO de dispositivos médicos, especialmente para las empresas que intentan pasar de conjuntos de capacidades limitadas hacia una entrega de extremo a extremo. La adquisición por parte de NAMSA de las operaciones de pruebas de dispositivos médicos de WuXi AppTec en los Estados Unidos en febrero de 2025 amplió la capacidad de laboratorio y la amplitud de los servicios, mientras que su acuerdo de enero de 2026 para el negocio de pruebas de dispositivos médicos de desarrollo temprano de Labcorp fortaleció la profundidad de las pruebas en microbiología, biocompatibilidad y áreas relacionadas. NAMSA también afirma que más del 70% de los estudios globales de biocompatibilidad son realizados por su organización, lo que muestra cuán concentrados se han vuelto ya algunos nichos críticos para el cumplimiento, aunque el mercado de CRO de dispositivos médicos en general siga siendo más disperso.
Otros grandes proveedores también están ampliando su alcance a través de movimientos de cartera específicos en lugar de depender únicamente de la expansión orgánica. ICON reveló su adquisición de KCR S.A. Group por USD 92,5 millones en 2024, y en diciembre de 2025 anunció un acuerdo para adquirir ClinicalRM con el fin de profundizar su posición en la investigación patrocinada por el gobierno y el trabajo en enfermedades infecciosas que también puede respaldar programas de dispositivos. La integración completa de Medidata Clinical Data Studio por parte de ICON en marzo de 2025 también apunta a un camino de diferenciación basado en tecnología, ya que la plataforma integra datos de fuentes de Medidata y no Medidata en un único flujo de trabajo para la gestión de datos, la revisión y el monitoreo central. Estos pasos muestran que el mercado de CRO de dispositivos médicos no compite únicamente en número de empleados o alcance geográfico, porque la profundidad de la plataforma y la integración del flujo de trabajo se están convirtiendo en parte de la oferta comercial.
La tecnología se está convirtiendo en un límite competitivo más visible en el mercado de CRO de dispositivos médicos porque los patrocinadores quieren cada vez más un socio que pueda trabajar en revisión de datos, monitoreo, registros de calidad y documentación regulatoria sin entregas repetidas. Al mismo tiempo, los proveedores especializados siguen siendo importantes porque el trabajo con dispositivos incluye categorías limitadas pero esenciales, como la biocompatibilidad, los extractables y lixiviables, y la validación centrada en la ciberseguridad, que las empresas generalistas más grandes no siempre controlan internamente. El mercado está, por tanto, viendo dos caminos a la vez, con grandes empresas que se expanden a través de adquisiciones e integración, mientras que las empresas de nicho defienden su posición a través de la profundidad técnica y una respuesta más rápida en líneas de servicio específicas. El espacio en blanco sigue siendo más visible donde los dispositivos con gran contenido de software requieren soporte alineado en evidencia clínica, manejo de datos con conciencia de privacidad y mantenimiento de calidad del ciclo de vida. Esto mantiene al mercado de CRO de dispositivos médicos competitivo, activo y solo moderadamente concentrado a pesar del papel más fuerte de algunos actores de escala en ciertos nichos de pruebas.
Líderes de la Industria de CRO de Dispositivos Médicos
IQVIA
NAMSA
Medpace
Charles River Laboratories
Avania
- *Nota aclaratoria: los principales jugadores no se ordenaron de un modo en especial

Desarrollos Recientes de la Industria
- Febrero de 2026: El QMSR de la FDA (21 CFR Parte 820 según enmienda) entró en vigor el 2 de febrero, incorporando la norma ISO 13485:2016 por referencia, retirando la Técnica de Inspección del Sistema de Calidad (QSIT) en favor de un proceso de inspección actualizado bajo el Programa de Cumplimiento 7382.850. El QMSR introduce la integración obligatoria de la gestión de riesgos a lo largo del ciclo de vida del producto y nuevos requisitos de mantenimiento de registros vinculados al UDI, ampliando la demanda de CRO para servicios de evaluación de brechas del sistema de gestión de calidad y revalidación.
- Enero de 2026: Labcorp anunció la venta de su negocio de pruebas de dispositivos médicos de Desarrollo Temprano a NAMSA. La transacción amplía la capacidad de pruebas de NAMSA en microbiología, biocompatibilidad y extractables/lixiviables, reforzando su posición como la CRO de pruebas preclínicas dominante en el sector de dispositivos médicos.


Alcance del Informe del Mercado Global de CRO de Dispositivos Médicos
Según el alcance del informe, una Organización de Investigación por Contrato (CRO) de dispositivos médicos es una empresa especializada que proporciona servicios externalizados a los fabricantes de dispositivos médicos. Estos servicios apoyan el desarrollo, las pruebas y la aprobación regulatoria de los dispositivos médicos. Las CRO ayudan a agilizar los ensayos clínicos, las presentaciones regulatorias y las actividades de cumplimiento, lo que permite a los fabricantes llevar sus dispositivos al mercado de manera más eficiente.
El mercado de CRO de dispositivos médicos está segmentado por escala de operación en fase preclínica/de pruebas y fase clínica. Por tipo de servicio, el mercado se categoriza en servicios de monitoreo clínico, servicios de gestión de ensayos clínicos, servicios de gestión de datos y bioestadística, servicios de asuntos regulatorios y médicos, servicios de redacción médica, servicios de reclutamiento de pacientes y sitios, servicios de seguridad y farmacovigilancia, y otros servicios. Por clase de dispositivo, la segmentación incluye dispositivos MedTech, dispositivos de diagnóstico, dispositivos portátiles y otros dispositivos. Por geografía, el mercado se divide en la región de América del Norte, la región europea, la región de Asia-Pacífico, la región de Oriente Medio y África, y la región de América del Sur. El informe de mercado también cubre los tamaños de mercado estimados y las tendencias para 17 países en las principales regiones a nivel mundial. Para cada segmento, el tamaño del mercado y el pronóstico se proporcionan en términos de valor (USD).
| Preclínico/Pruebas |
| Clínico |
| Monitoreo Clínico |
| Gestión de Ensayos Clínicos |
| Gestión de Datos y Bioestadística |
| Asuntos Regulatorios y Médicos |
| Redacción Médica |
| Reclutamiento de Pacientes y Sitios |
| Seguridad y Farmacovigilancia |
| Otros (Habilitación de Ensayos Clínicos Descentralizados y Plataformas eClinical, Imagen/Laboratorio Central, etc.) |
| Dispositivos MedTech |
| Dispositivos de diagnóstico |
| Dispositivos portátiles |
| Otros |
| América del Norte | Estados Unidos |
| Canadá | |
| México | |
| Europa | Alemania |
| Reino Unido | |
| Francia | |
| Italia | |
| España | |
| Resto de Europa | |
| Asia-Pacífico | China |
| India | |
| Japón | |
| Corea del Sur | |
| Australia | |
| Resto de Asia-Pacífico | |
| Oriente Medio y África | CCG |
| Sudáfrica | |
| Resto de Oriente Medio y África | |
| América del Sur | Brasil |
| Argentina | |
| Resto de América del Sur |
| Por Escala de Operación | Preclínico/Pruebas | |
| Clínico | ||
| Por Tipo de Servicio | Monitoreo Clínico | |
| Gestión de Ensayos Clínicos | ||
| Gestión de Datos y Bioestadística | ||
| Asuntos Regulatorios y Médicos | ||
| Redacción Médica | ||
| Reclutamiento de Pacientes y Sitios | ||
| Seguridad y Farmacovigilancia | ||
| Otros (Habilitación de Ensayos Clínicos Descentralizados y Plataformas eClinical, Imagen/Laboratorio Central, etc.) | ||
| Por Clase de Dispositivo | Dispositivos MedTech | |
| Dispositivos de diagnóstico | ||
| Dispositivos portátiles | ||
| Otros | ||
| Por Geografía | América del Norte | Estados Unidos |
| Canadá | ||
| México | ||
| Europa | Alemania | |
| Reino Unido | ||
| Francia | ||
| Italia | ||
| España | ||
| Resto de Europa | ||
| Asia-Pacífico | China | |
| India | ||
| Japón | ||
| Corea del Sur | ||
| Australia | ||
| Resto de Asia-Pacífico | ||
| Oriente Medio y África | CCG | |
| Sudáfrica | ||
| Resto de Oriente Medio y África | ||
| América del Sur | Brasil | |
| Argentina | ||
| Resto de América del Sur | ||


Preguntas Clave Respondidas en el Informe
¿Qué está impulsando el crecimiento en los servicios de CRO de dispositivos médicos hasta 2031?
El crecimiento está siendo respaldado por una mayor carga regulatoria, mayores necesidades de evidencia poscomercialización y el movimiento de USD 11,91 mil millones en 2026 a USD 16,82 mil millones en 2031 a una CAGR del 7,14%.
¿Qué área de servicio lidera la demanda externalizada para los patrocinadores de dispositivos?
Los servicios clínicos lideran con una participación del 58,46% en 2025, y también son el segmento de escala de operación de más rápido crecimiento con una CAGR del 7,67% hasta 2031.
¿Por qué los asuntos regulatorios y médicos crecen más rápido que otras líneas de servicio?
El QMSR, el MDR y el IVDR han aumentado la cantidad de documentación, alineación de calidad y trabajo de cumplimiento del ciclo de vida, razón por la cual se proyecta que los Asuntos Regulatorios y Médicos crezcan al 7,82% hasta 2031.
¿Qué clase de dispositivo crea la mayor oportunidad de externalización hoy en día?
Los dispositivos de diagnóstico mantuvieron la mayor participación con el 42,64% en 2025 porque el IVDR ha ampliado las necesidades de evidencia y documentación para una mayor proporción de la categoría.
¿Qué región se está expandiendo más rápido para el trabajo de desarrollo de dispositivos externalizado?
Se prevé que Asia-Pacífico crezca más rápido al 8,32% hasta 2031, respaldada por la creciente actividad de desarrollo regional y la necesidad de ejecución regulatoria localizada.
¿Qué tan concentrada está la competencia entre los proveedores de CRO de dispositivos médicos?
La competencia es moderada en lugar de altamente concentrada, porque las empresas globales de servicio completo están activas, pero muchos proveedores especializados todavía mantienen posiciones importantes en pruebas, trabajo regulatorio y validación digital.
Última actualización de la página el: