Taille et part du marché de la thérapie génique
VUE D’ENSEMBLE DU MARCHÉ
| Période d'étude | 2020 - 2031 |
|---|---|
| Taille du Marché (2026) | 10.04 Milliards de dollars |
| Taille du Marché (2031) | 25.89 Milliards de dollars |
| Taux de croissance (2026 - 2031) | 20.86% CAGR |
| Marché à la Croissance la Plus Rapide | Asie-Pacifique |
| Plus Grand Marché | Amérique du Nord |
| Concentration du Marché | Moyen |
Acteurs majeurs *Avis de non-responsabilité : les principaux acteurs sont triés sans ordre particulier Image © Mordor Intelligence. La réutilisation nécessite une attribution sous CC BY 4.0. | |
Analyse du marché de la thérapie génique par Mordor Intelligence
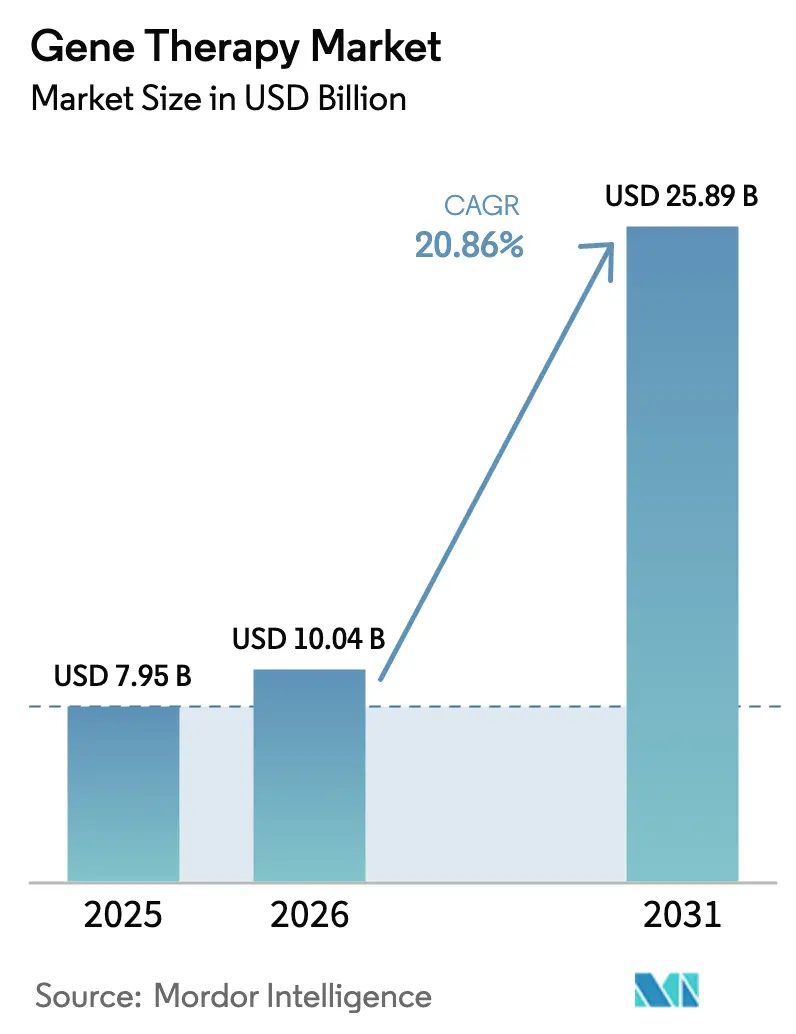
La taille du marché de la thérapie génique était évaluée à 7,95 milliards USD en 2025 et devrait croître de 10,04 milliards USD en 2026 pour atteindre 25,89 milliards USD d'ici 2031, à un TCAC de 20,86 % au cours de la période de prévision (2026-2031).
L'accélération des approbations de thérapies curatives à dose unique, les modèles de remboursement basés sur les résultats et les gains d'efficacité dans la fabrication de plateformes transforment la thérapie génique d'une modalité expérimentale en une classe de traitement commercialement évolutive. La réticence des payeurs s'atténue à mesure que les données du monde réel démontrent un bénéfice durable, tandis que les procédés standardisés de virus adéno-associé (AAV) et de lentivirus (LV) compriment les délais de production jusqu'à 50 %, soutenant des taux de succès des lots plus élevés. Les investisseurs en capital-risque se sont orientés vers des actifs en phase avancée, réduisant le risque de développement et dirigeant les capitaux vers des actifs à potentiel de revenus plus proche. Parallèlement, les systèmes de nanoparticules lipidiques non viraux arrivent à maturité, préparant une concurrence multiplateforme susceptible d'élargir davantage la portée thérapeutique. La concurrence en matière de propriété intellectuelle autour des capside modifiées et des charges utiles d'édition s'intensifie, plus de 500 brevets de capside AAV novateurs ayant été déposés entre 2020 et 2024, signalant une course décisive pour le leadership en matière d'administration.
Principaux enseignements du rapport
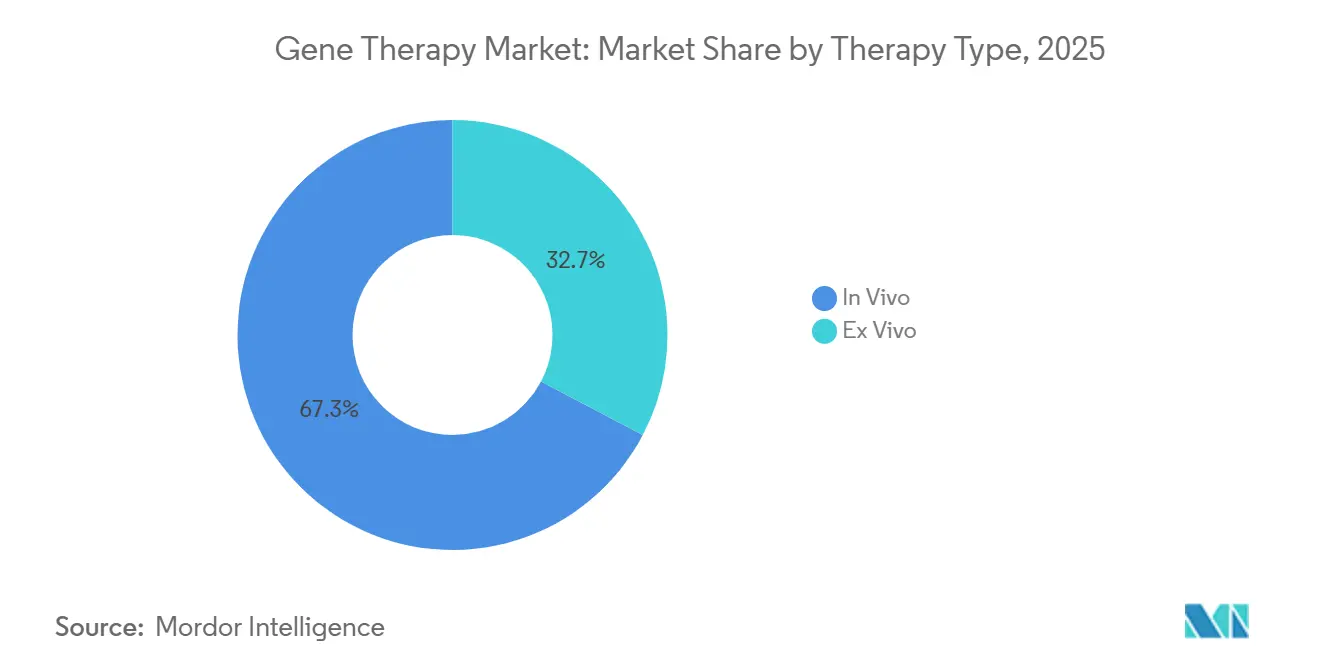
- Par type de thérapie, les approches in vivo ont dominé avec une part de revenus de 67,31 % en 2025, tandis que l'ex vivo affiche la croissance la plus rapide avec un TCAC de 21,97 % prévu jusqu'en 2031.
- Par type de vecteur, les vecteurs viraux représentaient 74,83 % de la part du marché de la thérapie génique en 2025 ; cependant, les systèmes non viraux devraient se développer à un TCAC de 23,41 % jusqu'en 2031.
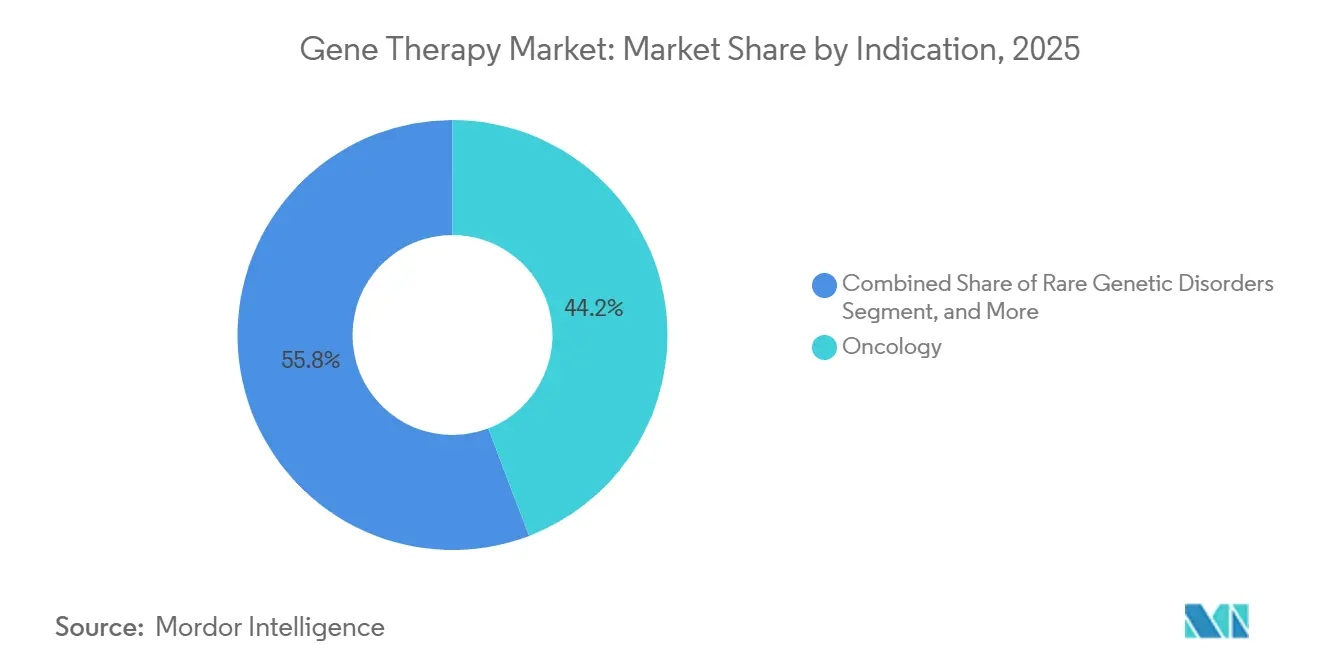
- Par indication, l'oncologie a généré 44,15 % des revenus de 2025, tandis que les applications en neurologie progressent à un TCAC de 22,71 % vers 2031.
- Par méthode d'administration, l'administration systémique représentait 46,36 % des ventes de 2025, tandis que les voies localisées devraient croître à 25,18 % jusqu'en 2031.
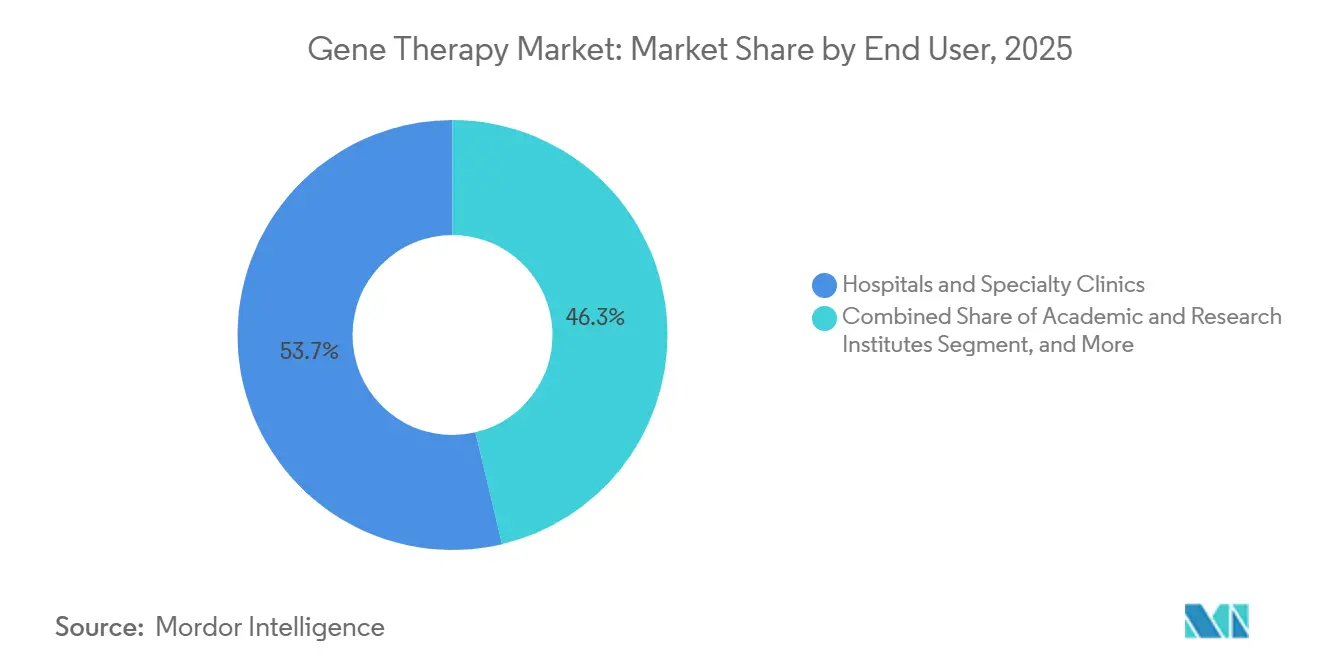
- Par utilisateur final, les hôpitaux et les cliniques spécialisées représentaient 53,66 % des revenus en 2025, tandis que les instituts académiques et de recherche sont en voie d'atteindre un TCAC de 26,64 % jusqu'en 2031.
- Par géographie, l'Amérique du Nord a conservé 41,36 % de la valeur de 2025, mais l'Asie-Pacifique est positionnée pour le TCAC le plus élevé de 28,78 % entre 2026 et 2031.
Note : La taille du marché et les prévisions figurant dans ce rapport sont générées à l'aide du cadre d'estimation exclusif de Mordor Intelligence, mis à jour avec les dernières données et informations disponibles en janvier 2026.
Tendances et Analyses du Marché
Analyse de l'impact des moteurs du marché de la thérapie génique*
| Moteur | (~) % d'impact sur la prévision du TCAC | Pertinence géographique | Horizon temporel de l'impact |
|---|---|---|---|
| Hausse des approbations de thérapies curatives à dose unique | +5.2% | Mondial, avec l'Amérique du Nord et l'Europe en tête | Moyen terme (2-4 ans) |
| Expansion du remboursement pour les maladies ultra-rares | +4.8% | Amérique du Nord, marchés européens sélectifs, émergent en Asie-Pacifique | Moyen terme (2-4 ans) |
| Standardisation des plateformes de fabrication (AAV et LV) | +4.1% | Mondial, concentré dans les pôles de fabrication d'Amérique du Nord, d'Europe et d'Asie de l'Est | Long terme (≥ 4 ans) |
| Croissance des pipelines de financement par capital-risque et SPAC | +2.9% | Amérique du Nord et Europe, avec des retombées vers Israël et Singapour | Court terme (≤ 2 ans) |
| Avancées dans l'édition in vivo basée sur CRISPR | +3.6% | Mondial, avec un leadership clinique aux États-Unis, au Royaume-Uni et en Suisse | Moyen terme (2-4 ans) |
| Inclusion croissante des thérapies géniques pour les maladies rares | +2.7% | Mondial, avec un fort soutien réglementaire en Amérique du Nord, dans l'UE et au Japon | Moyen terme (2-4 ans) |
| Source: Mordor Intelligence | |||
Hausse des approbations de thérapies curatives à dose unique
Les autorités réglementaires ont accéléré les délais en 2024-2025, autorisant des thérapies telles que Waskyra pour le syndrome de Wiskott-Aldrich, Itvisma pour l'amyotrophie spinale et Casgevy pour la drépanocytose dans le cadre de procédures prioritaires.[1]U.S. Food and Drug Administration, "Produits de thérapie cellulaire et génique approuvés," fda.gov Les données cliniques sous-tendant ces décisions démontrent une efficacité durable, Casgevy maintenant 93,5 % des patients sans crise sur 12 mois et Beqvez maintenant une activité du facteur IX supérieure à 40 %, éliminant ainsi le besoin de prophylaxie contre l'hémophilie B. Les payeurs reconnaissent que les interventions à dose unique peuvent compenser des décennies de coûts de soins chroniques, déclenchant des modèles budgétaires recalibrés. L'extension de la voie Sakigake du Japon à la thérapie génique souligne l'alignement plus large dans la région Asie-Pacifique, renforçant le virage mondial vers des paradigmes curatifs.
Expansion du remboursement pour les maladies ultra-rares
Les modèles basés sur les résultats régissent désormais environ 40 % des contrats commerciaux américains, et le modèle d'accès aux thérapies cellulaires et géniques du CMS couvre 35 programmes Medicaid, partageant le risque financier avec les fabricants.[2]Centers for Medicare & Medicaid Services, "Modèle d'accès aux thérapies cellulaires et géniques," cms.gov L'Europe reste fragmentée mais expérimente les paiements échelonnés, la HAS française étalant le coût de 2,1 millions USD de Zolgensma sur cinq ans. Les prévisions américaines du Congressional Budget Office indiquent que les thérapies contre la drépanocytose pourraient à elles seules générer 1,8 milliard USD de dépenses annuelles Medicaid d'ici 2028, incitant les législateurs à développer des solutions d'achat groupé. Les données de registre issues de l'utilisation réelle de Zolgensma indiquent que 95 % des enfants traités conservent leurs jalons moteurs à un suivi de cinq ans, renforçant la valeur des régimes de paiement à la performance.
Standardisation des plateformes de fabrication (AAV et LV)
L'installation de 1 milliard USD de Lonza à Portsmouth et l'expansion de 675 millions USD de Catalent à Harmans sont spécialement conçues pour des bioréacteurs modulaires allant de 50 à 2 000 litres, pouvant pivoter entre plusieurs campagnes sans nécessiter de transfert de procédé.[3]Lonza, "Lonza annonce une installation de 1 milliard USD à Portsmouth," lonza.com Le système AAV-MAX de MilliporeSigma réduit de moitié les coûts en amont en éliminant les exigences de culture adhérente, tandis que la plateforme Pro10™ de Viralgen a réduit les taux d'échec des lots à moins de 5 % sur 1 500 cycles. Samsung Biologics ajoute la première grande installation AAV d'Asie-Pacifique, préfigurant une chaîne d'approvisionnement géographiquement diversifiée. Le guide conjoint FDA-EMA de 2024 sur l'analytique AAV harmonise les tests de libération, réduisant directement les coûts de validation dupliqués.
Croissance des pipelines de financement par capital-risque et SPAC
Les investisseurs en capital-risque ont déployé 3,2 milliards USD en 2024, avec 70 % des fonds alloués aux actifs en Phase 2 et au-delà, indiquant une demande plus élevée de réduction du risque clinique. La Série C de 315 millions USD de Prime Medicine et la Série A de 213 millions USD de Tome Biosciences illustrent des paris surdimensionnés sur l'édition différenciée. L'activité SPAC a diminué en 2024-2025 alors que les marchés publics privilégiaient la visibilité des revenus ; cependant, les bras de capital-risque d'entreprise tels que Novo Holdings et Takeda Ventures sont restés actifs, apportant une expertise réglementaire et en CMC aux côtés du capital.
Analyse de l'impact des freins du marché de la thérapie génique*
| Frein | (~) % d'impact sur la prévision du TCAC | Pertinence géographique | Horizon temporel de l'impact |
|---|---|---|---|
| Coût des marchandises vendues élevé et réaction face aux prix à six chiffres | -3.7% | Mondial, aigu en Europe et dans les marchés émergents | Moyen terme (2-4 ans) |
| Mandats complexes de surveillance de la sécurité à long terme | -2.4% | Mondial, les plus stricts en Amérique du Nord et dans l'UE | Long terme (≥ 4 ans) |
| Goulots d'étranglement mondiaux dans la capacité de vecteurs viraux | -2.8% | Mondial, impact concentré dans les pôles de fabrication d'Amérique du Nord et d'Europe | Court terme (≤ 2 ans) |
| Émergence de fourrés de brevets sur les charges utiles d'édition génique | -1.9% | Mondial, le plus aigu en Amérique du Nord et en Europe avec des retombées vers l'Asie-Pacifique | Moyen terme (2-4 ans) |
| Source: Mordor Intelligence | |||
Coût des marchandises vendues élevé et réaction face aux prix à six chiffres
Les lots de vecteurs viraux coûtent entre 500 000 et 2 millions USD, entraînant des prix catalogue tels que les 4,25 millions USD de Lenmeldy, provoquant des auditions de la Commission des finances du Sénat américain et des rejets du NICE en dessous des seuils de 100 000 GBP/QALY. La variabilité des rendements allant de 30 % à 70 % par campagne oblige à la surproduction et aux dépréciations. Les pilotes de fabrication en continu de Resilience et Fujifilm Diosynth promettent une réduction des coûts de 50 % mais n'atteindront pas une échelle commerciale significative avant 2027. Le retrait de Zynteglo de l'UE par Bluebird en 2024, suite à des échecs de remboursement, illustre le risque existentiel posé par la réticence des organismes d'évaluation des technologies de santé à accepter les prix.
Mandats complexes de surveillance de la sécurité à long terme
La FDA exige une surveillance des patients sur 15 ans pour les vecteurs intégratifs, incluant des visites annuelles à triennales, ainsi qu'un dépistage des malignités hématologiques, ce qui coûte aux petites entreprises entre 5 et 10 millions USD par an. Les plans de gestion des risques de l'EMA prolongent davantage la charge jusqu'à l'obtention de l'autorisation de mise sur le marché complète, retardant ainsi les délais de rentabilité. Le REMS de Zolgensma et la surveillance de la leucémie de Lyfgenia illustrent les engagements post-commercialisation sur plusieurs décennies qui pèsent sur les développeurs à actif unique. L'initiative de registre volontaire de l'ASGCT pourrait centraliser les données mais manque de participation universelle.
*Nos prévisions considèrent les impacts des moteurs et des contraintes comme directionnels et non additifs. Les prévisions d'impact reflètent la croissance de référence, les effets de composition et les interactions entre variables.
Analyse des segments du marché de la thérapie génique
Par type de thérapie :
les plateformes ex vivo s'accélèrent grâce au potentiel allogéniqueLa taille du marché de la thérapie génique pour les approches in vivo représentait 67,31 % des revenus en 2025, reflétant sa domination dans les applications hépatiques et systémiques. Les thérapies ex vivo représentaient le reste mais devraient dépasser le marché à un TCAC de 21,97 %, alimentées par des conceptions de CAR-T allogéniques activées par CRISPR qui contournent les goulots d'étranglement autologues. Allogene et CRISPR Therapeutics ont avancé des essais de Phase 2 en 2024, démontrant des flux de travail veine-à-veine rapides de 14 jours. Le guide CMC provisoire de la FDA a clarifié les critères de test des lots et de dépistage des donneurs, réduisant l'ambiguïté réglementaire.
L'adoption dépend d'une fabrication rentable au point de soins. Lonza et Miltenyi Biotec ont piloté des salles blanches mobiles en 2024 qui réduisent les coûts par dose de 40 %, positionnant les sites hospitaliers comme des micro-usines. Les programmes in vivo rencontrent encore des problèmes d'immunogénicité ; jusqu'à 50 % des candidats possèdent des anticorps neutralisants, ce qui incite au développement de capsides modifiées. Les maladies neurodégénératives, menées par l'AMT-130 d'uniQure, montrent des promesses là où l'ex vivo est impraticable, maintenant une diversité thérapeutique élevée.



Par type de vecteur :
les systèmes non viraux comblent les lacunes d'efficacitéLes plateformes virales ont généré 74,83 % des revenus de 2025, ancrées par le tropisme hépatocytaire de l'AAV et l'intégration du LV pour les maladies hématologiques. Les vecteurs non viraux devraient se développer à un TCAC de 23,41 %, réduisant l'écart grâce à des nanoparticules lipidiques optimisées qui ont délivré des taux d'édition de 40 % à 60 % dans les modèles précliniques de Moderna et BioNTech. L'électroporation de MaxCyte soutient plus de 50 essais cliniques, offrant une expression transitoire adaptée à l'édition ex vivo.
Dans les catégories virales, les capsides AAV-PHP.B modifiées sont entrées dans des études de première administration chez l'homme pour la maladie de Parkinson, mettant en évidence le tropisme de nouvelle génération pour l'administration dans le système nerveux central. L'ADN enzymatique de Touchlight concurrence comme alternative aux plasmides évitant les contaminants bactériens, élargissant les options non virales. Le guide non viral de la FDA a standardisé les attentes en matière de qualité, alignant la surveillance sur le risque d'édition irréversible plutôt que sur la catégorie de vecteur.
Par indication :
la neurologie gagne en dynamismeLa contribution de 44,15 % de l'oncologie en 2025 devrait se poursuivre à mesure que les CAR-T progressent vers des lignes de traitement antérieures, Carvykti atteignant 73 % de réponses complètes dans le myélome multiple. Cependant, les indications en neurologie devraient croître à un TCAC de 22,71 % à mesure que l'administration intrathécale d'AAV contourne la barrière hémato-encéphalique. Les succès incluent la réduction de 79 % de la protéine huntingtine par uniQure et le programme GBA1 de Parkinson de Neurocrine. L'ophtalmologie reste un créneau rentable grâce au privilège immunitaire de l'œil, les données de durabilité sur cinq ans de Luxturna renforçant la confiance des payeurs.
Les maladies génétiques rares continuent de recevoir la priorité réglementaire, Waskyra et Itvisma obtenant des approbations en 2025. L'hématologie reste compétitive ; huit actifs contre l'hémophilie en développement en phase avancée pourraient comprimer le pouvoir de fixation des prix, motivant la différenciation par des doses de vecteurs plus faibles ou des contextes d'administration en ambulatoire.
Par méthode d'administration :
l'administration localisée s'intensifieLa perfusion systémique représentait 46,36 % des revenus en 2025, mais l'administration localisée est sur une trajectoire de TCAC de 25,18 %. Les approches intrathécales nécessitent un dixième de la dose de vecteur pour l'amyotrophie spinale, réduisant le risque immunogène. Les protocoles chirurgicaux sous-rétiniens standardisés par Luxturna sont désormais en opération dans 150 centres américains, garantissant des résultats reproductibles. L'administration intravitréenne est à l'étude pour la dégénérescence maculaire liée à l'âge humide, réduisant potentiellement les injections mensuelles d'anti-VEGF à des calendriers annuels. Les schémas systémiques dominent encore l'édition des hépatocytes mais font face à l'exclusion en raison du développement d'anticorps neutralisants, incitant à une reconception continue de la capside.
Par utilisateur final :
les instituts académiques propulsent l'innovation précoceLes hôpitaux et les cliniques spécialisées ont capturé 53,66 % des revenus de 2025, fonctionnant comme des pôles d'administration commerciale équipés de suites d'aphérèse BPF. Les instituts académiques, disposant d'une base plus petite, affichent un TCAC de 26,64 %, car 60 % des essais actifs proviennent de protocoles initiés par des investigateurs. Les réseaux européens de référence illustrent l'harmonisation transfrontalière, regroupant les cas de maladies rares pour des études à plus grande puissance. Les investissements hospitaliers tels que l'usine de traitement cellulaire de 50 millions USD du Massachusetts General renforcent la fabrication nationale, soutenant la production décentralisée au point de soins.
Analyse géographique
Marché de la thérapie génique en Amérique du Nord
L'Amérique du Nord a maintenu 41,36 % des revenus de 2025, soutenue par 45 nouvelles désignations RMAT et le modèle d'accès CMS facilitant les obstacles liés à Medicaid. Les capacités construites par Lonza, Catalent et Resilience ont totalisé 1,2 million de litres de bioréacteurs, consolidant la domination de la région en matière d'approvisionnement. Les contrats basés sur les résultats couvrent désormais 40 % des thérapies américaines, comme en témoigne la couverture de Lyfgenia pour 100 millions de vies. Le Canada est en retrait avec seulement deux approbations en 2024, en raison de négociations prolongées sur le remboursement provincial.
Marché de la thérapie génique en Asie-Pacifique
La région Asie-Pacifique, dont la croissance est projetée à un CAGR de 28,78 %, est portée par les approbations de Roctavian et Yescarta par la NMPA chinoise, ainsi que par plus de 200 essais cliniques actifs. L'installation de plasmides de WuXi AppTec et l'usine AAV de Samsung diversifient la fabrication mondiale. La PMDA japonaise a étendu la voie rapide Sakigake à la thérapie génique, et l'Inde a approuvé son premier CAR-T à un dixième du prix américain, illustrant l'innovation en matière de coûts. L'Australie a approuvé Casgevy mais fait face à des retards de remboursement.
Marché de la thérapie génique en EMEA et en Amérique du Sud
L'Europe est confrontée à des systèmes d'évaluation des technologies de santé fragmentés. Le retrait de Zynteglo par Bluebird à la suite d'échecs de remboursement illustre le risque commercial associé à l'approbation de l'EMA. L'évaluation positive de Hemgenix par l'IQWiG en Allemagne et les paiements échelonnés de la France pour Zolgensma constituent des succès isolés. Les évaluations conjointes 2025 d'EUnetHTA visent à harmoniser les données probantes, mais les payeurs nationaux contrôlent toujours les prix. Le Moyen-Orient et l'Afrique, ainsi que l'Amérique du Sud, restent des marchés naissants mais développent des cadres réglementaires, les Émirats arabes unis et le Brésil ayant approuvé certaines thérapies pour un usage compassionnel ou en milieu hospitalier public.


Paysage concurrentiel
Le secteur de la thérapie génique reste modérément fragmenté ; Novartis, Gilead et Bristol Myers Squibb ont ensemble généré une valeur élevée grâce aux franchises CAR-T en 2024. Les grands groupes intégrés verticalement continuent d'agrandir leurs installations, tandis que les biotechs de plateforme accordent des licences sur les capsides ou les éditeurs plus largement. L'acquisition de Beam par Pfizer et l'acquisition de Voyager par Novartis mettent en évidence la consolidation autour de technologies d'administration différenciées.
La dynamique concurrentielle dans les maladies du système nerveux central implique moins de 10 candidats en phase avancée, laissant un espace blanc de besoins non satisfaits pour les innovateurs disposant de capsides pénétrant la barrière hémato-encéphalique. Les acteurs de CAR-T allogéniques, tels que CRISPR Therapeutics, Allogene et Cellectis, pourraient perturber les acteurs autologues établis si les données de Phase 3 confirment une efficacité et une sécurité comparables. Les brevets stimulent les alliances stratégiques ; les plus de 500 dépôts de capsides AAV depuis 2020 font pression sur les entrants pour négocier des licences ou risquer des litiges.
Les spécialistes non viraux Moderna, BioNTech et Intellia Therapeutics exploitent le savoir-faire en nanoparticules lipidiques issu des vaccins à ARNm pour délivrer des charges utiles d'édition, atteignant 40 % à 60 % d'édition hépatique dans des modèles précliniques. La différenciation technologique, notamment en termes de sélection du promoteur, de taille de la charge utile et de tropisme organique, dictera probablement les flux de transactions et les valorisations davantage que le simple nombre de pipelines.
Leaders du secteur de la thérapie génique
Amgen Inc.
Novartis AG
bluebird bio Inc.
Biogen Inc.
Gilead Sciences Inc.
- *Avis de non-responsabilité : les principaux acteurs sont triés sans ordre particulier
Entreprises couvertes dans ce rapport sur le marché de la thérapie génique
- Amgen
- Beam Therapeutics
- bluebird bio Inc.
- Bristol-Myers Squibb
- Biogen
- CRISPR Therapeutic
- Editas Medicine
- Freeline Therapeutics
- Gilead Sciences
- Intellia Therapeutics
- LogicBio Therapeutics
- MeiraGTx
- Mustang Bio
- Novartis
- Orchard Therapeutics
- Passage Bio
- Pfizer
- Regenxbio
- Sangamo Therapeutics
- Sarepta Therapeutics
- Spark Therapeutics
- Takeda Pharmaceuticals
- uniQure N.V.
- ViGeneron

Développements récents du secteur sur le marché de la thérapie génique
- Mai 2025 : Le MHLW japonais a approuvé ELEVIDYS (delandistrogene moxeparvovec-rokl) de Sarepta Therapeutics, une thérapie génique AAV à dose unique pour la dystrophie musculaire de Duchenne chez les enfants âgés de 3 à 7 ans sans délétions spécifiques du gène DMD et négatifs pour les anticorps anti-AAVrh74. Accordée dans le cadre d'une approbation conditionnelle et soutenue par les données de Phase 3 EMBARK, il s'agit de la première approbation mondiale couvrant les enfants de moins de 4 ans, avec Chugai et Roche gérant la commercialisation.
- Mai 2025 : Abeona Therapeutics a reçu l'approbation de la FDA pour ZEVASKYN (prademagene-zamifermin), la première thérapie génique autologue à base cellulaire pour les plaies d'épidermolyse bulleuse dystrophique récessive chez les adultes et les enfants. Les résultats de Phase 3 VIITAL ont démontré une cicatrisation durable des plaies et un soulagement de la douleur après une seule application, avec un lancement prévu au troisième trimestre 2025 via des centres spécialisés.
- Février 2025 : Genprex a consolidé ses licences de l'Université de Pittsburgh en un seul accord exclusif pour les technologies de thérapie génique du diabète utilisant les gènes Pdx1 et MafA. La société a également créé une filiale en propriété exclusive, Convergen Biotech, pour faire avancer GPX-002 dans le traitement du diabète de type 1 et de type 2, afin de mener des études préalables à l'IND d'ici fin 2025.
- Octobre 2024 : La gouverneure de New York Kathy Hochul a lancé la prochaine phase du New York BioGenesis Park, un pôle de thérapie cellulaire et génique de 430 millions USD sur Long Island, soutenu par un investissement record de l'État de 150 millions USD. L'installation vise à renforcer la recherche, la fabrication et la commercialisation, consolidant ainsi le leadership de New York dans les thérapies avancées.
- Septembre 2024 : Genprex a annoncé le transfert de son programme de thérapie génique du diabète, incluant GPX-002 pour le traitement du diabète de type 1 et de type 2, vers une nouvelle filiale en propriété exclusive (« NewCo »). Cette démarche sépare l'actif de reprogrammation des cellules alpha en cellules bêta du pipeline en oncologie afin d'accélérer le développement et d'obtenir un financement ciblé.


Marché de la thérapie génique Portée du rapport et méthodologie de recherche
Définitions du marché et périmètre de couverture
Notre étude définit le marché de la thérapie génique comme le chiffre d'affaires généré par des administrations uniques ou répétées qui insèrent, silencent ou modifient le matériel génétique à l'intérieur d'un patient, à l'aide de vecteurs viraux ou non viraux, afin de corriger des troubles héréditaires ou acquis dans l'ensemble des aires thérapeutiques et des zones géographiques.
Exclusion du périmètre : Les traitements qui se limitent à l'expansion ou à la réinfusion de cellules manipulées sans altération génomique directe (par exemple, les perfusions de cellules souches autologues) sont exclus.
Aperçu de la segmentation
- Par type de thérapie
- In Vivo
- Ex Vivo
- Par type de vecteur
- Vecteurs viraux
- Virus adéno-associé
- Lentivirus
- Adénovirus
- Rétrovirus et γ-rétrovirus
- Autres vecteurs viraux
- Vecteurs non viraux
- Vecteurs viraux
- Par indication
- Oncologie
- Maladies génétiques rares
- Ophtalmologie
- Hématologie
- Neurologie
- Cardiovasculaire et autres
- Par méthode d'administration
- Administration systémique
- Administration localisée
- Par utilisateur final
- Hôpitaux et cliniques spécialisées
- Instituts académiques et de recherche
- Autres utilisateurs finaux
- Par géographie
- Amérique du Nord
- États-Unis
- Canada
- Mexique
- Europe
- Allemagne
- Royaume-Uni
- France
- Italie
- Espagne
- Reste de l'Europe
- Asie-Pacifique
- Chine
- Japon
- Inde
- Australie
- Corée du Sud
- Reste de l'Asie-Pacifique
- Moyen-Orient et Afrique
- CCG
- Afrique du Sud
- Reste du Moyen-Orient et de l'Afrique
- Amérique du Sud
- Brésil
- Argentine
- Reste de l'Amérique du Sud
- Amérique du Nord
Méthodologie de recherche détaillée et validation des données
Recherche primaire
Les analystes de Mordor ont interrogé des examinateurs réglementaires, des pharmaciens hospitaliers spécialisés en thérapie génique, des dirigeants de CDMO vectoriels et des investigateurs cliniques en Amérique du Nord, en Europe et en Asie-Pacifique. Leurs éclairages ont permis d'affiner les données réelles sur le nombre de patients traités, les écarts entre prix catalogue et prix nets des thérapies, ainsi que les hypothèses de cadence de fabrication, ancrant ainsi des paramètres que la seule littérature ne peut révéler.
Recherche documentaire
Nous avons commencé par des sources de données ouvertes telles que la liste des approbations de thérapies cellulaires et géniques de la FDA américaine, clinicaltrials.gov, le registre des thérapies avancées de l'EMA et les statistiques de l'American Society of Gene & Cell Therapy, qui décrivent les lancements de thérapies, les volumes d'essais et les évolutions des capacités vectorielles. Les données financières sectorielles issues des 10-K déposés auprès de la SEC, des bases de données de remboursement des payeurs, des tableaux de bord de financement du NIH et des revues à comité de lecture ont fourni des informations sur les prix, la prévalence et la profondeur du pipeline, tandis que D&B Hoovers et Dow Jones Factiva nous ont aidés à estimer les chiffres d'affaires des entreprises et à suivre les flux de financement. Ces références sont données à titre illustratif uniquement ; de nombreuses sources supplémentaires ont contribué aux vérifications et clarifications des données.
Dimensionnement du marché et prévisions
Une reconstruction descendante du bassin de patients a mis en correspondance la prévalence des maladies avec les cohortes éligibles, lesquelles sont ensuite ajustées en fonction de l'adoption des thérapies, des extensions d'indication et de l'attrition, avant d'être multipliées par les prix réalisés mixtes. Des agrégations ascendantes sélectives des chiffres d'affaires publiquement divulgués pour les produits approuvés ont permis de vérifier les totaux. Les variables clés comprennent : (1) les approbations annuelles de la FDA/EMA, (2) les courbes de pénétration des patients traités, (3) le prix de vente net moyen par classe de vecteur, (4) le taux d'utilisation des capacités mondiales de fabrication d'AAV et de LNP, et (5) les dépenses de financement de la R&D. Nous projetons les valeurs 2025-2030 à l'aide d'une régression multivariée reliant ces facteurs aux trajectoires de chiffre d'affaires observées, et les lacunes dans les données ascendantes sont comblées par des plages de scénarios validées par nos interlocuteurs.
Cycle de validation des données et de mise à jour
Les résultats sont soumis à une révision par plusieurs analystes, à des tests de variance par rapport à des références externes et à des déclencheurs d'anomalies. Les rapports sont actualisés chaque année, avec des mises à jour intermédiaires lorsque des événements significatifs, tels que l'approbation d'une nouvelle thérapie à prix élevé, modifient la base de référence.
Pourquoi la base de référence de Mordor en thérapie génique est fiable
Les estimations publiées s'alignent rarement, car les entreprises retiennent des paniers de thérapies, des bases de devises et des cadences d'actualisation différents.
Nous mettons en évidence ces leviers dès le départ, afin que les clients sachent immédiatement ce qui est inclus ou non dans notre chiffre de 9,74 milliards USD pour 2025.
Comparaison avec les références
| Taille du marché | Source anonymisée | Principal facteur d'écart |
|---|---|---|
| 9,74 Md USD (2025) | Mordor Intelligence | - |
| 8,85 Md USD (2024) | Global Consultancy A | Année de base plus ancienne ; exclut les éditions in vivo gagnant des parts de marché |
| 11,07 Md USD (2025) | Industry Publisher B | Comptabilise le chiffre d'affaires des services CDMO et applique les prix catalogue |
Ces comparaisons montrent que lorsque l'élargissement du périmètre et l'optimisme tarifaire sont écartés, Mordor fournit une base de référence solide et transparente que les décideurs peuvent retracer jusqu'à des variables explicites et des étapes reproductibles.


Questions clés auxquelles le rapport répond
À quelle vitesse le marché de la thérapie génique devrait-il croître d'ici 2031 ?
Le secteur devrait se développer à un TCAC de 20,86 %, portant les revenus de 10,04 milliards USD en 2026 à 25,89 milliards USD d'ici 2031.
Quel type de thérapie devrait connaître la croissance la plus rapide ?
Les thérapies ex vivo affichent le TCAC le plus rapide de 21,97 %, portées par les programmes CAR-T allogéniques qui éliminent le besoin d'une fabrication spécifique au patient.
Pourquoi les payeurs commencent-ils à couvrir les thérapies géniques à prix élevé ?
Les contrats basés sur les résultats et les données du monde réel démontrant un bénéfice durable ont convaincu les payeurs que les coûts uniques peuvent compenser les dépenses de gestion des maladies à vie.
Quelle région est positionnée pour la croissance la plus élevée d'ici 2031 ?
L'Asie-Pacifique est en tête avec un TCAC projeté de 28,78 % alors que la Chine, le Japon et la Corée du Sud alignent leurs voies réglementaires et développent leur capacité de fabrication.
Qu'est-ce qui limite une adoption plus large malgré la dynamique réglementaire ?
Les coûts de fabrication élevés, les exigences de surveillance de la sécurité à long terme et les contraintes de capacité des vecteurs viraux créent des défis de coûts et d'approvisionnement qui tempèrent l'adoption.
Dernière mise à jour de la page le: