Marktgröße und Marktanteil für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen
Marktübersicht
| Studienzeitraum | 2020 - 2031 |
|---|---|
| Marktgröße (2026) | 9.37 Milliarden US-Dollar |
| Marktgröße (2031) | 14.19 Milliarden US-Dollar |
| Wachstumsrate (2026 - 2031) | 8.65% CAGR |
| Schnellstwachsender Markt | Asien-Pazifik |
| Größter Markt | Nordamerika |
| Marktkonzentration | Mittel |
Hauptakteure *Haftungsausschluss: Hauptakteure in keiner bestimmten Reihenfolge sortiert Bild © Mordor Intelligence. Wiederverwendung erfordert Namensnennung gemäß CC BY 4.0. | |
Marktanalyse für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen von Mordor Intelligence
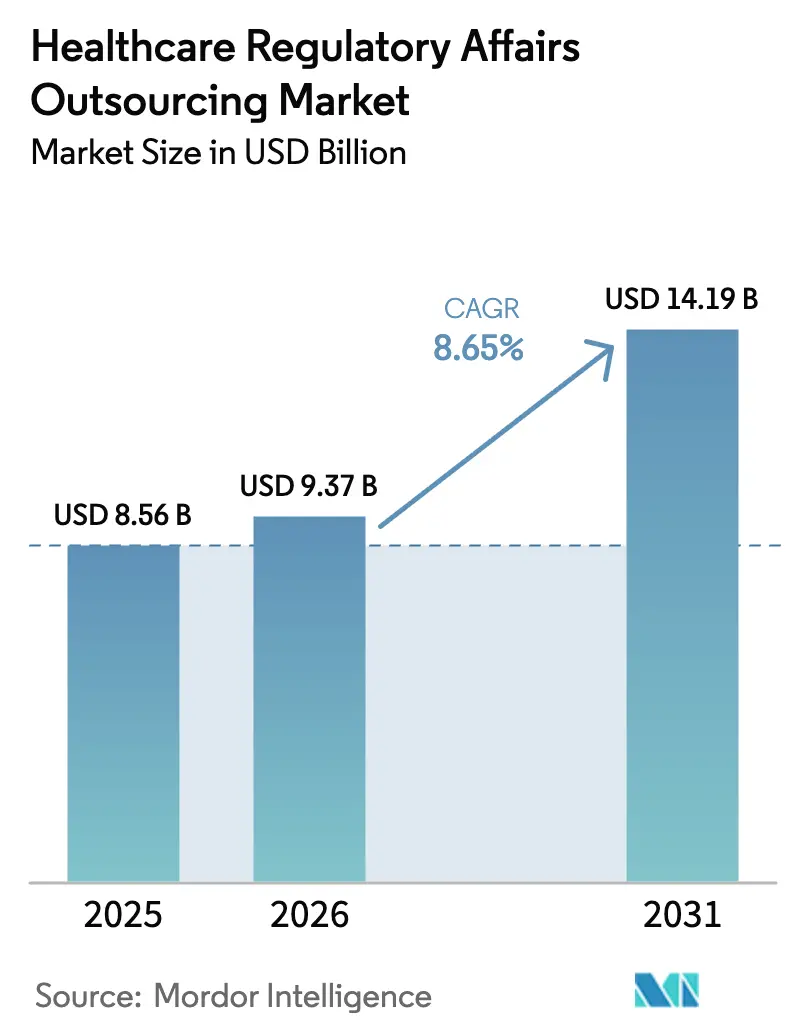
Die Marktgröße für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen wird voraussichtlich von 8,56 Milliarden USD im Jahr 2025 auf 9,37 Milliarden USD im Jahr 2026 wachsen und soll bis 2031 bei einer CAGR von 8,65 % über den Zeitraum 2026–2031 einen Wert von 14,19 Milliarden USD erreichen.
Der Wachstumspfad spiegelt die steigende Nachfrage virtueller Biotechnologieunternehmen wider, denen interne Regulierungskapazitäten fehlen, die Migration der umfangreichen Dossier-Erstellung zu spezialisierten Anbietern sowie immer strengere globale Einreichungsstandards. Auftraggeber priorisieren weiterhin die Markteinführungsgeschwindigkeit gegenüber dem Dokumentationsvolumen – eine Verschiebung, die erklärt, warum Produktregistrierung und klinische Studienanträge schneller wachsen als andere Dienstleistungen. Gleichzeitig verlangen Regulierungsbehörden kontinuierliche Real-World-Evidenz, wodurch Aktivitäten nach der Zulassung zu einem mehrjährigen Einnahmestrom für Auslagerungsanbieter werden. Nordamerika bleibt der Umsatzanker, doch der Aufstieg mehrsprachiger Zentren in Indien und China verändert die Wettbewerbslandschaft. Anbieter, die KI in Einreichungsabläufe integrieren, verkürzen Zykluszeiten, sichern sich größere Arbeitspakete und differenzieren sich durch Mehrwert statt durch Arbeitskosten.
Wichtigste Erkenntnisse des Berichts
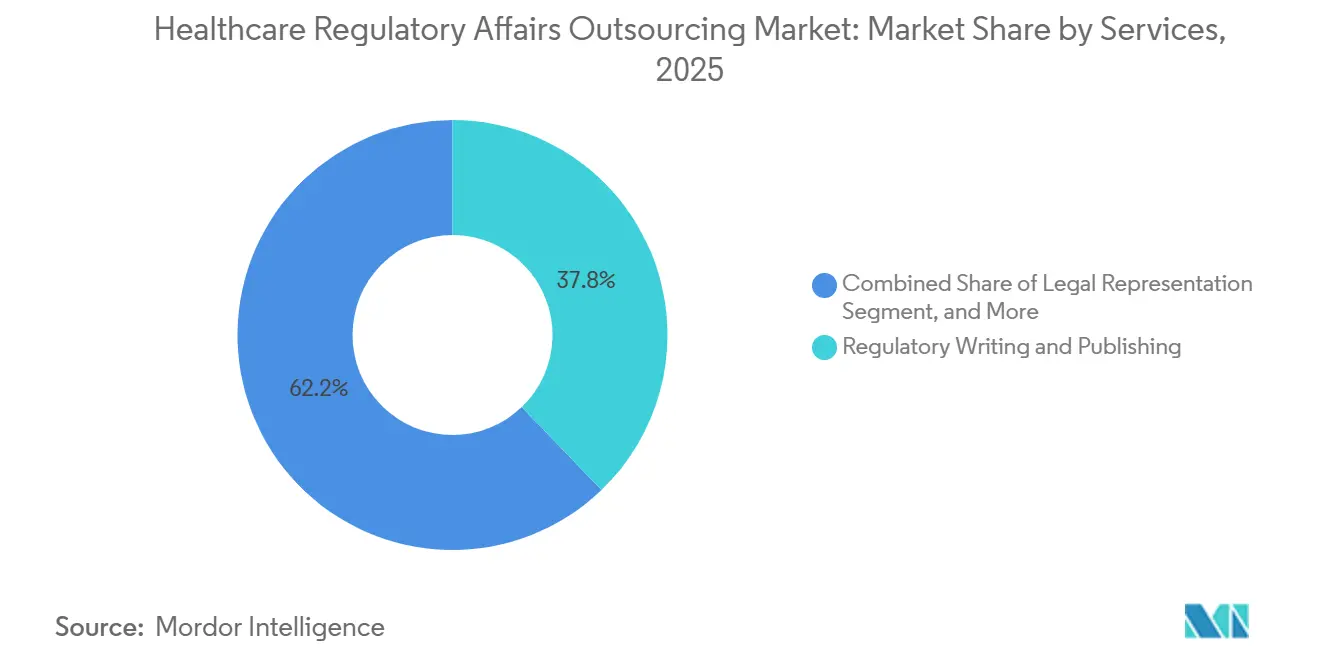
- Nach Dienstleistung führte das regulatorische Schreiben und Publizieren mit einem Umsatzanteil von 37,81 % im Jahr 2025; Produktregistrierung und klinische Studienanträge werden voraussichtlich bis 2031 mit einer CAGR von 11,66 % wachsen.
- Nach Produktlebenszyklusphase hielt die klinische Phase im Jahr 2025 einen Anteil von 44,73 % am Markt für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen, während Dienstleistungen nach der Zulassung und nach der Markteinführung bis 2031 mit einer CAGR von 12,42 % zunahmen.
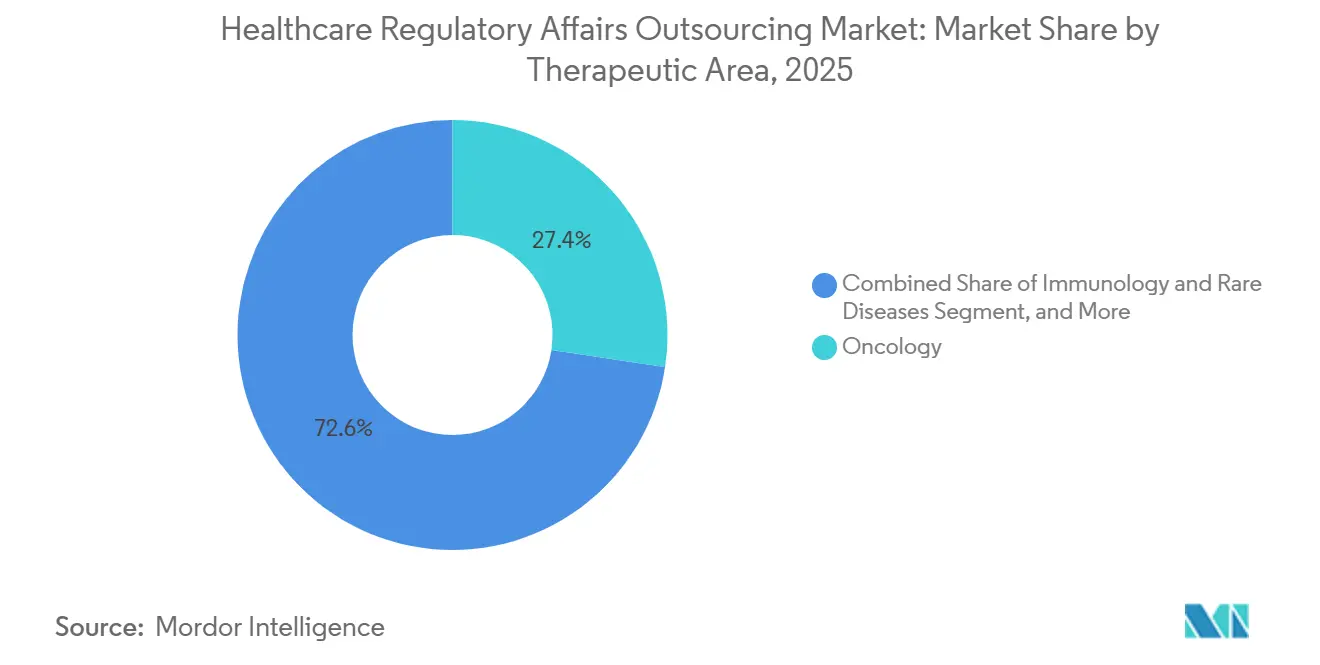
- Nach Therapiegebiet entfiel im Jahr 2025 ein Anteil von 27,38 % der Ausgaben auf die Onkologie; Immunologie und seltene Krankheiten verzeichneten mit einer CAGR von 10,19 % bis 2031 das schnellste Wachstum.
- Nach Endnutzer entfielen im Jahr 2025 58,36 % der Ausgaben auf Pharmaunternehmen, während Medizingerätehersteller die höchste prognostizierte CAGR von 9,36 % bis 2031 verzeichneten.
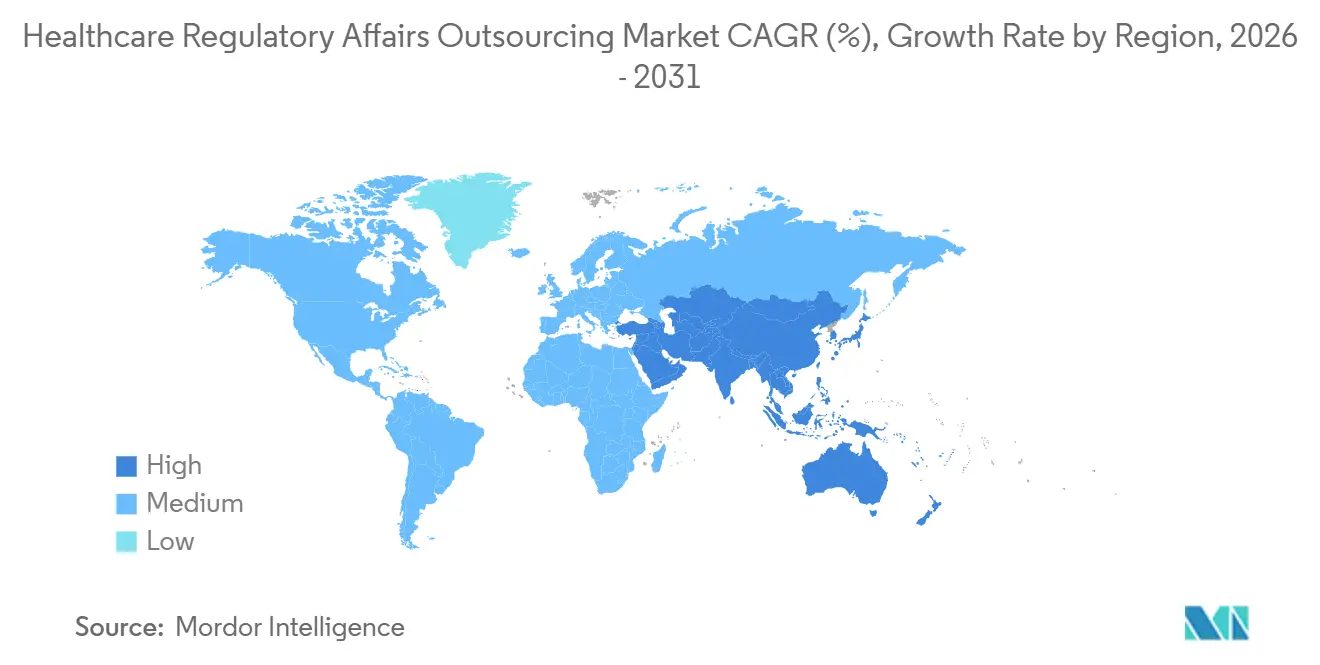
- Nach Geografie entfielen im Jahr 2025 42,36 % des Umsatzes auf Nordamerika; die Region Asien-Pazifik verzeichnet das schnellste Wachstum mit einer prognostizierten CAGR von 13,06 % bis 2031.
Hinweis: Die Marktgröße und Prognosezahlen in diesem Bericht werden mithilfe des proprietären Schätzungsrahmens von Mordor Intelligence erstellt und mit den neuesten verfügbaren Daten und Erkenntnissen vom Januar 2026 aktualisiert.
Globale Markttrends und Erkenntnisse zur Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen
Analyse der Auswirkungen von Treibern*
| Treiber | (~) % Auswirkung auf die CAGR-Prognose | Geografische Relevanz | Zeithorizont der Auswirkung |
|---|---|---|---|
| Steigende Anzahl klinischer Studien | +1.8% | Global, mit Schwerpunkt in Nordamerika, Europa und aufstrebenden Zentren im Asien-Pazifik-Raum | Mittelfristig (2–4 Jahre) |
| Fokussierung von Life-Science-Unternehmen auf Kernkompetenzen | +1.5% | Global, besonders stark in Nordamerika und Europa, wo virtuelle Biotechnologieunternehmen verbreitet sind | Langfristig (≥4 Jahre) |
| Wachsende Komplexität globaler regulatorischer Rahmenbedingungen | +1.4% | Global, akut in Märkten mit jüngsten regulatorischen Überarbeitungen (EU-MDR, Reformen der chinesischen NMPA) | Langfristig (≥4 Jahre) |
| Expansion virtueller/kleinmolekularer Biotech-Start-ups | +1.3% | Kernregionen Nordamerika und Europa, Ausstrahlungseffekte auf Israel und Singapur | Mittelfristig (2–4 Jahre) |
| KI-gestützte Übernahme von Regulierungsintelligenz | +1.2% | Frühe Anwender in Nordamerika und der EU, schrittweise Verbreitung im Asien-Pazifik-Raum | Kurzfristig (≤2 Jahre) |
| Entstehung kostengünstiger regulatorischer Zentren in Entwicklungsländern | +1.1% | Kernregion Asien-Pazifik (Indien, China, Philippinen), Ausstrahlungseffekte auf Osteuropa und Lateinamerika | Mittelfristig (2–4 Jahre) |
| Quelle: Mordor Intelligence | |||
Steigende Anzahl klinischer Studien
ClinicalTrials.gov verzeichnete bis Dezember 2024 487.000 Studien, was einem Anstieg von 6,2 % gegenüber 2023 entspricht, und fast die Hälfte der neuen Studien betraf Onkologie oder seltene Krankheiten.[1]National Library of Medicine, "ClinicalTrials.gov," CLINICALTRIALS.GOV Jede Studie erzeugt IND-Einreichungen, Protokolländerungen und Sicherheitsberichte, die Auftraggeber bevorzugt auslagern. Das Projekt Optimus der FDA verpflichtet Onkologieprogramme zur Aufnahme von Dosisoptimierungsstudien, was zusätzliche Dokumentationsebenen schafft, die kleine Teams belasten. Dezentralisierte Modelle umfassen mittlerweile rund 30 % der Phase-II- und Phase-III-Protokolle, die elektronische Einwilligung und Fernüberwachung kombinieren und regulatorische Unklarheiten einführen, die spezialisierte Anbieter besser bewältigen können. Die Nachfrage steigt auch nach Autoren, die digitale Biomarkerdaten in Dossiers integrieren können. Zusammen vergrößern diese Kräfte die Vertragswerte und stärken den Markt für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen.
Fokussierung von Life-Science-Unternehmen auf Kernkompetenzen
Virtuelle Biotechnologieunternehmen arbeiten häufig mit schlanker Belegschaft und lagern alle Compliance-Aufgaben von der Vor-IND-Phase bis zur Überwachung nach der Markteinführung aus. Etablierte Pharmaunternehmen behalten ihre Strategien intern, lagern jedoch umfangreiche Tätigkeiten aus, wie regionale Dossier-Anpassung, Kennzeichnungsüberarbeitungen und Serialisierungsaktualisierungen. Diese Zweiteilung veranlasst Anbieter, sowohl hochwertige Beratungsleistungen als auch standardisierte Dokumentenerstellung zu unterstützen. Gebührenstrukturen verlagern sich hin zu ergebnisbasierter Preisgestaltung, die an Zulassungsmeilensteine geknüpft ist und Anbieter belohnt, die Erstzyklus-Zulassungen erzielen können. Da Investoren die regulatorische Bereitschaft vor Finanzierungsrunden prüfen, engagieren Frühphasenunternehmen Berater früher und verankern die Auslagerung als strukturelle, nicht zyklische Entscheidung.
Wachsende Komplexität globaler regulatorischer Rahmenbedingungen
Die EU-Medizinprodukteverordnung, die 2024 vollständig umgesetzt wurde, erweiterte die Anforderungen an klinische Nachweise und Verpflichtungen nach der Markteinführung in allen 27 Mitgliedstaaten. Die chinesische Nationale Medizinproduktebehörde überarbeitete im selben Jahr ihr elektronisches Einreichungsformat und verschärfte Fristen sowie Datenintegritätsprüfungen. Das Real-World-Evidence-Programm der FDA genehmigt Beobachtungsdaten für Labelergänzungen, bietet jedoch begrenzte Designleitlinien, was Auftraggeber dazu veranlasst, Experten hinzuzuziehen, die vertretbare Methoden entwickeln können.[2]U.S. Food and Drug Administration, "AI/ML-Enabled Medical Devices," FDA.GOV Japan führte eine bedingte Zulassung für regenerative Medizin ein, was Langzeit-Follow-up-Studien erforderlich macht, die die Rolle der Anbieter weiter ausweiten. Überschneidende Rahmenbedingungen für Software als Medizinprodukt und KI-Gesundheitstools führen zusätzliche Compliance-Ebenen ein und verwandeln die regulatorische Navigation in einen spezialisierten Beruf, der in den meisten internen Gruppen oft nicht verfügbar ist.
KI-gestützte Übernahme von Regulierungsintelligenz
Der Entwurf der FDA-Leitlinie zu KI-gestützten Arzneimittelentwicklungstools, der im Mai 2024 veröffentlicht wurde, legitimierte Modelle des maschinellen Lernens für das Protokolldesign und die Vorhersage unerwünschter Ereignisse. Anbieter reagierten darauf, indem sie natürliche Sprachverarbeitung integrierten, die eCTD-Module automatisch zusammenstellt und Einreichungszyklen um bis zu 40 % verkürzt. Die Plattform Orchestrated Clinical Trials von IQVIA erkennt Compliance-Lücken während der Protokollerstellung und verlagert die Qualitätskontrolle in eine frühere Phase. Das IRIS-Portal der EMA erfordert strukturierte Dateneinreichungen und motiviert Auftraggeber, KI-Tools einzusetzen, die veraltete PDFs in regulierungskonforme Formate umwandeln. Trotz des Versprechens haben die Behörden noch keine endgültigen Validierungsstandards für KI-generierte Inhalte festgelegt, sodass Auftraggeber Berater mit doppelter Expertise in Datenwissenschaft und regulatorischen Angelegenheiten einstellen. Dieser Premium-Dienst treibt das Umsatzwachstum im Markt für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen voran.
Analyse der Auswirkungen von Hemmnissen*
| Hemmnis | (~) % Auswirkung auf die CAGR-Prognose | Geografische Relevanz | Zeithorizont der Auswirkung |
|---|---|---|---|
| Datensicherheits- und IP-Leckagenrisiken | -0.9% | Global, akut in Regionen mit grenzüberschreitenden Datenübertragungsbeschränkungen (EU-DSGVO, chinesisches PIPL) | Kurzfristig (≤2 Jahre) |
| Mangel an globaler Prozessstandardisierung | -0.7% | Global, besonders herausfordernd für multinationale Auftraggeber, die Einreichungen in mehreren Regionen verwalten | Langfristig (≥4 Jahre) |
| Steigende Kosten für spezialisiertes regulatorisches Fachpersonal | -0.5% | Nordamerika und Westeuropa, wo der Wettbewerb um erfahrene Fachkräfte intensiv ist | Mittelfristig (2–4 Jahre) |
| Schnelle politische Veränderungen bei digitalen Therapeutika | -0.4% | Nordamerika und EU, wo regulatorische Rahmenbedingungen für digitale Gesundheit noch reifen | Kurzfristig (≤2 Jahre) |
| Quelle: Mordor Intelligence | |||
Datensicherheits- und IP-Leckagenrisiken
Auftraggeber müssen vertrauliche Studiendaten und Formulierungsdetails an Dritte übermitteln; grenzüberschreitende Regelungen wie die DSGVO und das chinesische Gesetz zum Schutz personenbezogener Informationen verhängen jedoch hohe Bußgelder bei Verstößen. Ransomware-Vorfälle, die 2024 mehrere regulatorische Einreichungen zum Stillstand brachten, verdeutlichten die Anfälligkeit und erzeugten eine Nachfrage nach SOC-2-Typ-II-Zertifizierungen und jährlichen Penetrationstests. Der Verlust von geistigem Eigentum ist subtiler: Wenn ein Anbieter konkurrierende Auftraggeber in derselben Therapieklasse betreut, können strategische Erkenntnisse unbeabsichtigt weitergegeben werden. Vertragsverhandlungen umfassen nun detaillierte Cyber-Kontrollen, Datenresidenzklauseln und Prüfungsrechte, was Zeit und Kosten für Auslagerungsgeschäfte erhöht und das Segmentwachstum dämpft. Anbieter, die frühzeitig in Zero-Trust-Architekturen, dedizierte Clean Rooms und strenge Konfliktmanagementrichtlinien investieren, schützen ihren Umsatz und das Vertrauen ihrer Kunden.
Schnelle politische Veränderungen bei digitalen Therapeutika
Die FDA veröffentlichte 2024 11 Leitliniendokumente zu klinischer Entscheidungsunterstützung und Änderungskontrolle für KI-Geräte, die meisten davon bleiben jedoch Entwürfe, sodass Compliance-Ziele fließend bleiben. EU-Vorschriften klassifizieren viele digitale Therapeutika als Klasse-II-Medizinprodukte gemäß der Medizinprodukteverordnung; bis Ende 2024 gab es jedoch nur 23 benannte Stellen, was einen Zulassungsengpass schafft.[3]European Commission, "Medical Devices – New Regulations," EC.EUROPA.EU Erstattungswege unterscheiden sich je nach Land und schaffen kommerzielle Unsicherheit. Das Auslaufen der US-amerikanischen Durchsetzungsermessenspolitik, die während des COVID-19-Notstands die Vormarktzulassung für viele risikoarme Gesundheits-Apps ausgesetzt hatte, löste eine Welle nachträglicher Einreichungen aus, die sowohl Auftraggeber als auch Anbieter überwältigte. Da sich Rahmenbedingungen schneller weiterentwickeln, als interne Teams umgeschult werden können, beauftragen Kunden Berater nur für kurze, taktische Projekte, was mehrjährige Vertragswerte begrenzt und die Expansion des Marktes für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen hemmt.
*Unsere Prognosen behandeln die Auswirkungen von Treibern und Einschränkungen als richtungsweisend und nicht additiv. Die Wirkungsprognosen berücksichtigen Basiswachstum, Mischungseffekte und Wechselwirkungen zwischen Variablen.
Segmentanalyse
Nach Dienstleistungen: Dokumentenerstellung verankert den Umsatz, Markteinführungsgeschwindigkeit treibt das Wachstum
Regulatorisches Schreiben und Publizieren erzielte 2025 den größten Anteil von 37,81 % und unterstreicht die anhaltende Nachfrage nach konformen klinischen Studienberichten, Prüferinformationen und eCTD-Dossiers, die den ICH-Formatierungsregeln entsprechen. Produktregistrierung und klinische Studienanträge werden voraussichtlich bis 2031 mit einer CAGR von 11,66 % wachsen, da Auftraggeber Zeitpläne durch die Auslagerung paralleler Einreichungen in mehreren Regionen verkürzen möchten. Das eCTD-4.0-Mandat der FDA, das ab Januar 2025 gilt, hat die Auslagerung bei Unternehmen intensiviert, denen die erforderlichen Systeme oder geschultes Personal fehlen. Die Preise bleiben am höchsten für Regulierungsberatungsaufträge, die mit Modalitäten wie CRISPR-editierten Therapien verbunden sind, wo behördliche Präzedenzfälle selten sind und frühe wissenschaftliche Beratungssitzungen erfahrene Experten erfordern. Rechtliche Vertretung bleibt eine Nischen-, aber margenstarke Tätigkeit für Patent- und Exklusivitätsverhandlungen.
Kennzeichnung und Artwork-Management sind in der Größe bescheiden, wachsen aber stetig aufgrund von Serialisierungsmandaten und mehrsprachigen Verpackungsanforderungen. Aktivitäten nach der Markteinführung und Lebenszyklusmanagement steigen mit einer CAGR von 12,42 %, angetrieben durch die FDA-Sentinel-Initiative und die EudraVigilance-Anforderungen der EMA, die eine kontinuierliche Sicherheitsüberwachung vorschreiben. Anbieter, die Echtzeit-Sicherheitssignalerkennung und beschleunigte periodische Sicherheitsaktualisierungsberichte anbieten, gewinnen langfristige Verträge. Abonnementbasierte Regulierungsintelligenz, Lückenanalysen und Scheinprüfungen bilden sonstige Nischendienstleistungen, die sich während Prüfungszyklen oder beim Aufkommen neuer Leitlinien ausweiten. Der kumulative Effekt dieser Dienstleistungstrends erhält eine starke Dynamik im Markt für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen aufrecht.



Nach Produktlebenszyklusphase: Anstieg nach der Zulassung spiegelt regulatorische Wachsamkeit wider
Klinische Aktivitäten hielten 2025 einen Anteil von 44,73 % der Ausgaben und umfassten IND-Einreichungen, Protokolländerungen und Sicherheitsaktualisierungen. Aktivitäten nach der Zulassung und nach der Markteinführung werden bis 2031 voraussichtlich mit einer CAGR von 12,42 % wachsen, da Regulierungsbehörden Bestätigungsstudien für beschleunigte Zulassungen vorschreiben und strengere Pharmakovigilanzstandards einführen. Die FDA erteilte 2024 15 Onkologiemedikamenten eine beschleunigte Zulassung, von denen jedes Studien nach der Markteinführung erfordert, um die Nachfrage nach externer regulatorischer Unterstützung aufrechtzuerhalten. Registrierungsprojekte erreichen ihren Höhepunkt kurz vor der Erstzulassung, umfassen nun aber iterative Interaktionen, da Behörden rollierende Überprüfungen einführen. Die Strategie der EMA für regulatorische Wissenschaft betont adaptive Wege und verpflichtet Auftraggeber, den Dialog auch nach der Dossiereinreichung aufrechtzuerhalten. Die Marktgröße für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen für Verpflichtungen nach der Zulassung wird daher voraussichtlich schneller wachsen als die traditionelle klinische Auslagerung im Prognosezeitraum.
Präklinische Beratung ist zwar kleiner, zeigt aber einen Aufwärtstrend, da Auftraggeber frühzeitiges Behördenfeedback schätzen und kostspielige Neugestaltungen später vermeiden möchten. Die kontinuierliche Generierung von Real-World-Evidenz verwischt die Grenzen zwischen Aktivitäten nach der Markteinführung und klinischen Aktivitäten, was Kunden dazu veranlasst, integrierte Anbieter zu bevorzugen, die Produkte über ihren gesamten Lebenszyklus begleiten können.
Nach Therapiegebiet: Seltene Krankheiten überholen die etablierte Führungsposition der Onkologie
Die Onkologie machte 2025 27,38 % der Ausgaben aus, da Kombinationsregime und biomarkergesteuerte Studien die Einreichungen komplexer machten. Immunologie und seltene Krankheiten, begünstigt durch 301 Orphan-Drug-Designierungen im Jahr 2024, werden voraussichtlich mit einer CAGR von 10,19 % wachsen. Surrogatendpunkte, die im Rahmen von Orphan- und Breakthrough-Programmen akzeptiert werden, verkürzen Zeitpläne, erfordern aber differenzierte Nutzen-Risiko-Berichte, die nur wenige interne Mitarbeiter erstellen können. Infektionskrankheitsprogramme erhielten neuen Auftrieb durch den Anreiz für qualifizierte Infektionskrankheitsprodukte, was das Dokumentationsvolumen erneut vergrößerte.
Kardiometabolische Therapien erweitern die Indikationen für GLP-1-Agonisten auf Adipositas und Lebererkrankungen und erzeugen statistische Komplexitäten, die Auftraggeber zu externen Biostatistikern und Autoren drängen. ZNS und Neurologie bleibt nach mehreren Alzheimer-Rückschlägen hochriskant, doch jedes Programm, das Phase III erreicht, erfordert nun intensive Beratungsengagements, um Endpunkte mit den Erwartungen der Behörden abzustimmen. Gen- und Zelltherapien erzielen Premiumgebühren, da Anbieter Vergleichbarkeits-, Potenz- und Langzeitsicherheitsfragen ansprechen müssen, die für diese Modalitäten einzigartig sind. Insgesamt stellen diese Dynamiken sicher, dass die therapeutische Segmentierung weiterhin die Tiefe im Markt für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen vorantreibt.
Nach Endnutzer: Gerätehersteller beschleunigen sich, während Pharma dominiert
Pharmaunternehmen machten 2025 58,36 % des Umsatzes aus und spiegeln ihre großen Pipelines und komplexen Einreichungen in mehreren Regionen wider. Medizingerätehersteller werden bis 2031 voraussichtlich eine CAGR von 9,36 % verzeichnen, da die EU-Medizinprodukteverordnung und die FDA-Regeln zur eindeutigen Geräteidentifikation zusätzliche Dokumentationsanforderungen einführen. Biotechnologieunternehmen, die häufig virtuell operieren, lagern nahezu alle regulatorischen Aufgaben aus, und ihre Anzahl wächst weiter, da Risikokapitalinvestitionen anhalten. Auftragsforschungs- und Auftragsfertigungsorganisationen betten regulatorische Dienstleistungen in schlüsselfertige Angebote ein und komprimieren die Margen für eigenständige Boutiquen.
Kombinationsprodukte wie Arzneimittel-Gerät-Autoinjektoren verwischen die Zuständigkeitsgrenze zwischen CDER und CDRH und machen duale Wegexpertise sehr wertvoll. Geräteunternehmen, die eine De-novo-Klassifizierung anstreben, verlassen sich auf externe Strategen, um analytische und klinische Daten vorzulegen, die den Standards der angemessenen Sicherheit entsprechen. Die kumulative Komplexität über alle Endnutzer hinweg unterstützt eine nachhaltige Expansion im Markt für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen.
Geografische Analyse
Nordamerika machte 2025 42,36 % des Umsatzes aus, unterstützt durch mehr als 5.000 aktive Biotech-Start-ups und die Position der FDA als globale Referenzregulierungsbehörde. Die Region Asien-Pazifik wird voraussichtlich mit einer CAGR von 13,06 % wachsen, was die politischen Reformen Indiens und die aggressive Digitalisierung von Einreichungen in China widerspiegelt, die Eintrittsbarrieren für lokale Anbieter senken. Die indische Zentrale Drogen-Standardkontrollorganisation genehmigte 2024 62 neue Arzneimittelanträge, was einem Anstieg von 28 % entspricht und eine verbesserte Prüfungseffizienz signalisiert. China bearbeitete im selben Jahr mehr als 1.100 Arzneimittelregistrierungen, 80 % von inländischen Akteuren, was die lokale Beratungsnachfrage weiter ankurbelt.
Der Nahe Osten und Afrika beschleunigen sich, da Mitglieder des Golfkooperationsrats Arzneimittelpreise und Registrierungsprozesse harmonisieren und damit die Einreichungsvariabilität verringern. Südamerika bleibt fragmentiert, wobei die ANVISA-Zeitpläne Brasiliens von denen der argentinischen ANMAT abweichen, was regionalen Unternehmen ermöglicht, eine dominante Position zu behalten. Gegenseitige Anerkennungsabkommen zwischen der FDA und Regulierungsbehörden in der EU, Kanada und Australien rationalisieren Inspektionen und begünstigen globale Auftragsforschungsorganisationen mit standardisierten Prozessen. Diese regionalen Kontraste prägen die Expansionsstrategien der Anbieter im Markt für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen.

Wettbewerbslandschaft
Der Markt für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen bleibt mäßig fragmentiert. Full-Service-Auftragsforschungsorganisationen wie IQVIA, Charles River Laboratories und ICON integrieren regulatorische Angelegenheiten in End-to-End-Entwicklungspakete und nutzen operative Daten, um Einreichungen zu beschleunigen. Die Übernahme von PPD durch Thermo Fisher Scientific für 17,4 Milliarden USD signalisierte den Wert interner regulatorischer Talente für diversifizierte Life-Science-Anbieter. Kleinere Beratungsunternehmen differenzieren sich durch Spezialisierung auf seltene Krankheiten, Zell- und Gentherapien oder KI-gestützte Einreichungsplattformen.
Die Übernahme von Technologien ist der wichtigste Wettbewerbshebel. ICON und Microsoft nutzen generative KI, um die eCTD-Kompilierungszeit um 35 % zu reduzieren. Die PPD-Division von Thermo Fisher lancierte ein Dashboard für Regulierungsintelligenz, das Aktualisierungen von 50 Behörden aggregiert und prädiktive Prüfungszeitpläne liefert, die Auftraggeber schätzen. Anbieter ohne digitale Tools stehen unter Margendruck, da Kunden schnellere Zykluszeiten zu niedrigeren Kosten fordern. Fusionen und Übernahmen zielen auf Boutique-Unternehmen mit geografischer Tiefe oder Software-Assets ab, die die Dokumentenverarbeitung rationalisieren. Trotz Konsolidierung bestehen Marktlücken bei digitalen Therapeutika und der Beratung zu Software als Medizinprodukt – Bereiche mit wenigen erfahrenen Beratern, aber steigendem Einreichungsvolumen – was die Wettbewerbsintensität stabil hält.
Marktführer im Bereich der Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen
IQVIA
Parexel International Corporation
ICON PLC
Charles River Laboratories
Labcorp
- *Haftungsausschluss: Hauptakteure in keiner bestimmten Reihenfolge sortiert


Jüngste Branchenentwicklungen
- Januar 2026: Medispend und RLDatix Life Sciences haben ihre Fusion abgeschlossen und eine einheitliche Organisation gebildet, die unter dem Namen Medispend operieren wird. Das kombinierte Unternehmen bietet nun eine umfassende Suite von Unternehmenssoftware und -dienstleistungen an, die globale regulatorische und kommerzielle Compliance, medizinische Angelegenheiten, Außendienstunterstützung und Umsatzmanagemenlösungen abdeckt. Dieser strategische Schritt soll Life-Science-Unternehmen in die Lage versetzen, ihr Geschäft auszubauen und gleichzeitig die Compliance in verschiedenen Märkten aufrechtzuerhalten.
- Oktober 2025: Die PPD-Division von Thermo Fisher Scientific hat eine KI-gestützte Plattform für Regulierungsintelligenz eingeführt, die Echtzeit-Aktualisierungen von 50 globalen Gesundheitsbehörden aggregiert, darunter FDA, EMA, PMDA und NMPA. Die Plattform nutzt maschinelles Lernen, um Prüfungszeitpläne vorherzusagen und potenzielle Compliance-Lücken zu erkennen, und bietet Auftraggebern ein proaktives Tool zur Risikominderung bei Einreichungen und zur Vermeidung kostspieliger Änderungen.
- Januar 2025: ICON plc hat eine Partnerschaft mit Microsoft geschlossen, um generative KI-Tools für die eCTD-Kompilierung einzusetzen und die Dokumentenzusammenstellungszeit um schätzungsweise 35 % zu reduzieren. Die Zusammenarbeit integriert den Azure OpenAI Service von Microsoft in die regulatorischen Arbeitsabläufe von ICON und ermöglicht die automatisierte Erstellung von klinischen Studienberichten und Prüferinformationen, die den ICH-Formatierungsstandards entsprechen.


Umfang des globalen Berichts über den Markt für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen
Die Auslagerung regulatorischer Angelegenheiten umfasst Dienstleistungen, die von Pharma-, Biotech- und Medizingeräteherstellern genutzt werden, um schnelle regulatorische Zulassungen von verschiedenen Organisationen zu erhalten und Kosten zu sparen. Der Markt für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen ist segmentiert nach Dienstleistungen (Regulierungsberatung, Rechtliche Vertretung, Regulatorisches Schreiben und Publizieren, Produktregistrierung und klinische Studienanträge sowie sonstige Dienstleistungen), Endnutzer (Pharma- und Biotechnologieunternehmen sowie Medizingeräteunternehmen) und Geografie (Nordamerika, Europa, Asien-Pazifik, Naher Osten und Afrika sowie Südamerika). Der Marktbericht umfasst auch die geschätzten Marktgrößen und Trends für 17 verschiedene Länder in den wichtigsten Regionen weltweit. Der Bericht bietet den Wert (in USD) für die oben genannten Segmente.
| Regulierungsberatung |
| Rechtliche Vertretung |
| Regulatorisches Schreiben und Publizieren |
| Produktregistrierung und klinische Studienanträge |
| Kennzeichnung und Artwork-Management |
| Aktivitäten nach der Markteinführung und Lebenszyklusmanagement |
| Sonstige Nischendienstleistungen |
| Präklinisch |
| Klinisch (Phase I–III) |
| Registrierung |
| Nach der Zulassung und nach der Markteinführung |
| Onkologie |
| Infektionskrankheiten |
| Kardiometabolisch |
| ZNS und Neurologie |
| Immunologie und seltene Krankheiten |
| Sonstige Therapiegebiete |
| Pharmaunternehmen |
| Biotechnologieunternehmen |
| Medizingerätehersteller |
| Auftragsforschungs- und Auftragsfertigungsorganisationen |
| Nordamerika | Vereinigte Staaten |
| Kanada | |
| Mexiko | |
| Europa | Deutschland |
| Vereinigtes Königreich | |
| Frankreich | |
| Italien | |
| Spanien | |
| Übriges Europa | |
| Asien-Pazifik | China |
| Japan | |
| Indien | |
| Australien | |
| Südkorea | |
| Übriger Asien-Pazifik-Raum | |
| Naher Osten und Afrika | Golfkooperationsrat |
| Südafrika | |
| Übriger Naher Osten und Afrika | |
| Südamerika | Brasilien |
| Argentinien | |
| Übriges Südamerika |
| Nach Dienstleistungen | Regulierungsberatung | |
| Rechtliche Vertretung | ||
| Regulatorisches Schreiben und Publizieren | ||
| Produktregistrierung und klinische Studienanträge | ||
| Kennzeichnung und Artwork-Management | ||
| Aktivitäten nach der Markteinführung und Lebenszyklusmanagement | ||
| Sonstige Nischendienstleistungen | ||
| Nach Produktlebenszyklusphase | Präklinisch | |
| Klinisch (Phase I–III) | ||
| Registrierung | ||
| Nach der Zulassung und nach der Markteinführung | ||
| Nach Therapiegebiet | Onkologie | |
| Infektionskrankheiten | ||
| Kardiometabolisch | ||
| ZNS und Neurologie | ||
| Immunologie und seltene Krankheiten | ||
| Sonstige Therapiegebiete | ||
| Nach Endnutzer | Pharmaunternehmen | |
| Biotechnologieunternehmen | ||
| Medizingerätehersteller | ||
| Auftragsforschungs- und Auftragsfertigungsorganisationen | ||
| Nach Geografie | Nordamerika | Vereinigte Staaten |
| Kanada | ||
| Mexiko | ||
| Europa | Deutschland | |
| Vereinigtes Königreich | ||
| Frankreich | ||
| Italien | ||
| Spanien | ||
| Übriges Europa | ||
| Asien-Pazifik | China | |
| Japan | ||
| Indien | ||
| Australien | ||
| Südkorea | ||
| Übriger Asien-Pazifik-Raum | ||
| Naher Osten und Afrika | Golfkooperationsrat | |
| Südafrika | ||
| Übriger Naher Osten und Afrika | ||
| Südamerika | Brasilien | |
| Argentinien | ||
| Übriges Südamerika | ||


Im Bericht beantwortete Schlüsselfragen
Wie hoch ist der aktuelle Wert des Marktes für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen?
Der Markt für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen hat im Jahr 2026 einen Wert von 9,37 Milliarden USD.
Wie hoch ist die prognostizierte Wachstumsrate des Marktes für die Auslagerung regulatorischer Angelegenheiten im Gesundheitswesen?
Es wird prognostiziert, dass er mit einer CAGR von 8,65 % wächst und bis 2031 einen Wert von 14,19 Milliarden USD erreicht.
Welches Dienstleistungssegment erzielt den höchsten Umsatz?
Regulatorisches Schreiben und Publizieren führt mit einem Anteil von 37,81 % am Umsatz 2025.
Welche Region wird das schnellste Wachstum verzeichnen?
Asien-Pazifik wird bis 2031 voraussichtlich mit einer CAGR von 13,06 % wachsen.
Warum gewinnen Dienstleistungen nach der Zulassung an Bedeutung?
Regulierungsbehörden fordern nun kontinuierliche Real-World-Evidenz und eine intensivierte Pharmakovigilanz, was eine CAGR von 12,42 % bei der Auslagerung nach der Markteinführung antreibt.
Seite zuletzt aktualisiert am: