Alport-Syndrom Marktgröße und Marktanteil
Marktübersicht
| Studienzeitraum | 2020 - 2031 |
|---|---|
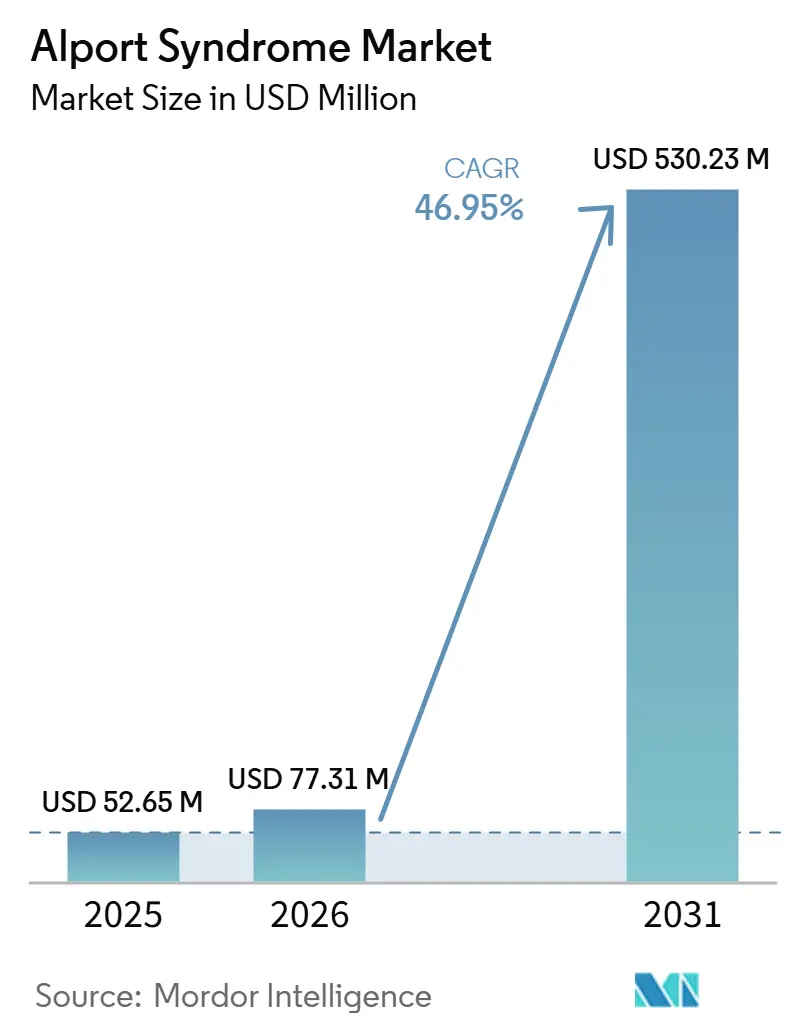
| Marktgröße (2026) | 77.31 Millionen US-Dollar |
| Marktgröße (2031) | 530.23 Millionen US-Dollar |
| Wachstumsrate (2026 - 2031) | 46.95% CAGR |
| Schnellstwachsender Markt | Asien-Pazifik |
| Größter Markt | Nordamerika |
| Marktkonzentration | Mittel |
Hauptakteure *Haftungsausschluss: Hauptakteure in keiner bestimmten Reihenfolge sortiert Bild © Mordor Intelligence. Wiederverwendung erfordert Namensnennung gemäß CC BY 4.0. | |
Alport-Syndrom Marktanalyse von Mordor Intelligence
Die Größe des Alport-Syndrom-Marktes wird voraussichtlich von 52,65 Millionen USD im Jahr 2025 auf 77,31 Millionen USD im Jahr 2026 steigen und bis 2031 530,23 Millionen USD erreichen, mit einem CAGR von 46,95 % über den Zeitraum 2026–2031.
Der Alport-Syndrom-Markt erzielt den größten Teil seiner aktuellen Einnahmen nach wie vor aus unterstützender Pharmakotherapie, Nierenersatzleistungen und genetischer Diagnostik, da keine Therapie speziell für diese Erkrankung eine FDA- oder EMA-Zulassung besitzt. Der Alport-Syndrom-Markt befindet sich nun in einer anderen Phase, da mindestens 4 Prüfmoleküle in der Phase-2- oder Phase-2b-Entwicklung aktiv waren und pivotale Studien voraussichtlich vor 2027 beginnen werden.[1]Bayer AG, "Bayer startet Phase-IIa-Studie zur Behandlung von Patienten mit Alport-Syndrom," Bayer AG Pressemitteilung, bayer.com Die erste zugelassene krankheitsmodifizierende Therapie im Alport-Syndrom-Markt würde ohne einen direkt zugelassenen Konkurrenten eintreten, und diese Position würde durch Rahmenbedingungen für Orphan-Drug-Exklusivität in wichtigen Rechtsordnungen gestärkt. Der Alport-Syndrom-Markt weitet sich auch durch familiäres Kaskaden-Screening aus, da variantenspezifische Tests bei gefährdeten Verwandten den diagnostizierten Pool vergrößern, ohne die tatsächliche Krankheitsinzidenz zu verändern. Die Durchführung von Studien bleibt komplex, da das RAAS-Ansprechen je nach Genotyp unterschiedlich ist und eine schwächere Proteinuriereduktion bei Fällen mit trunkierenden COL4A5-Varianten dazu führt, dass sich der Alport-Syndrom-Markt in Richtung einer stärker stratifizierten Therapieentwicklung bewegt.
Wichtigste Erkenntnisse des Berichts
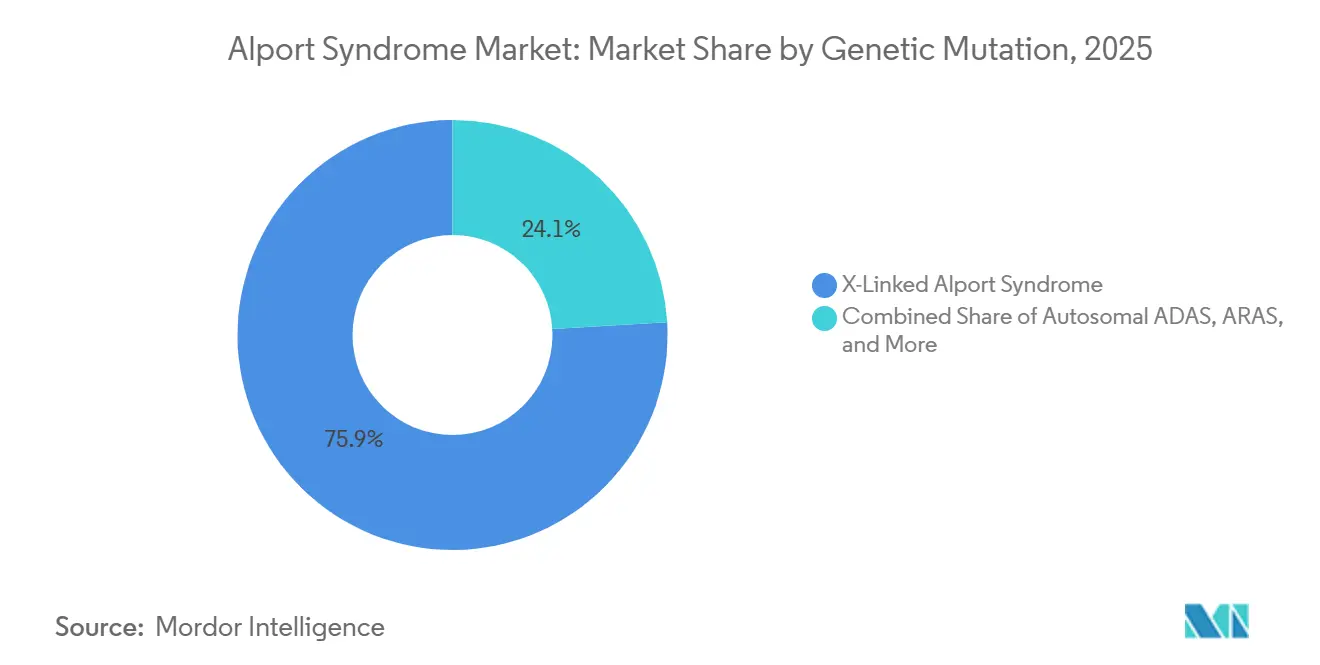
Nach genetischer Mutation hielt das X-chromosomale Alport-Syndrom im Jahr 2025 einen Marktanteil von 75,94 %, während das autosomal-dominante Alport-Syndrom bis 2031 voraussichtlich mit einem CAGR von 52,87 % wachsen wird.
Nach Behandlungsart entfiel im Jahr 2025 ein Anteil von 65,11 % auf die unterstützende pharmakologische Therapie, während die Nierenersatztherapie bis 2031 voraussichtlich mit einem CAGR von 55,34 % expandieren wird.
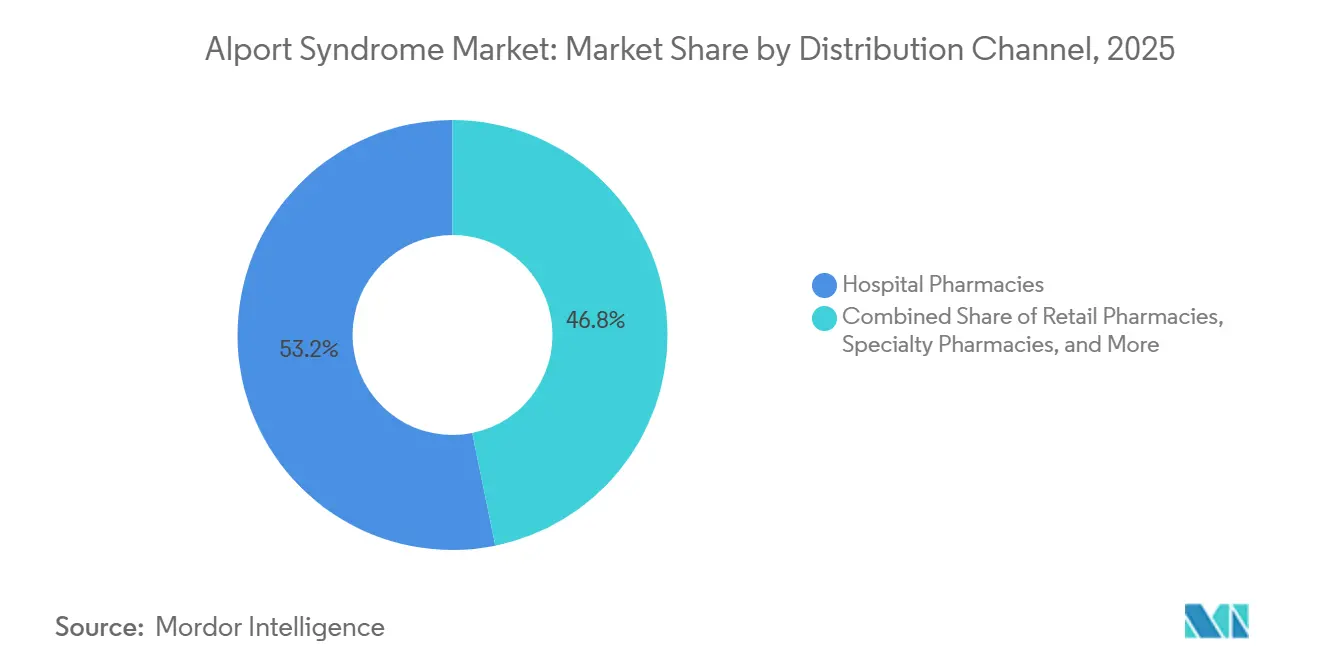
Nach Vertriebskanal repräsentierten Krankenhausapotheken im Jahr 2025 einen Anteil von 53,18 %, während Spezialapotheken bis 2031 voraussichtlich mit einem CAGR von 53,17 % wachsen werden.
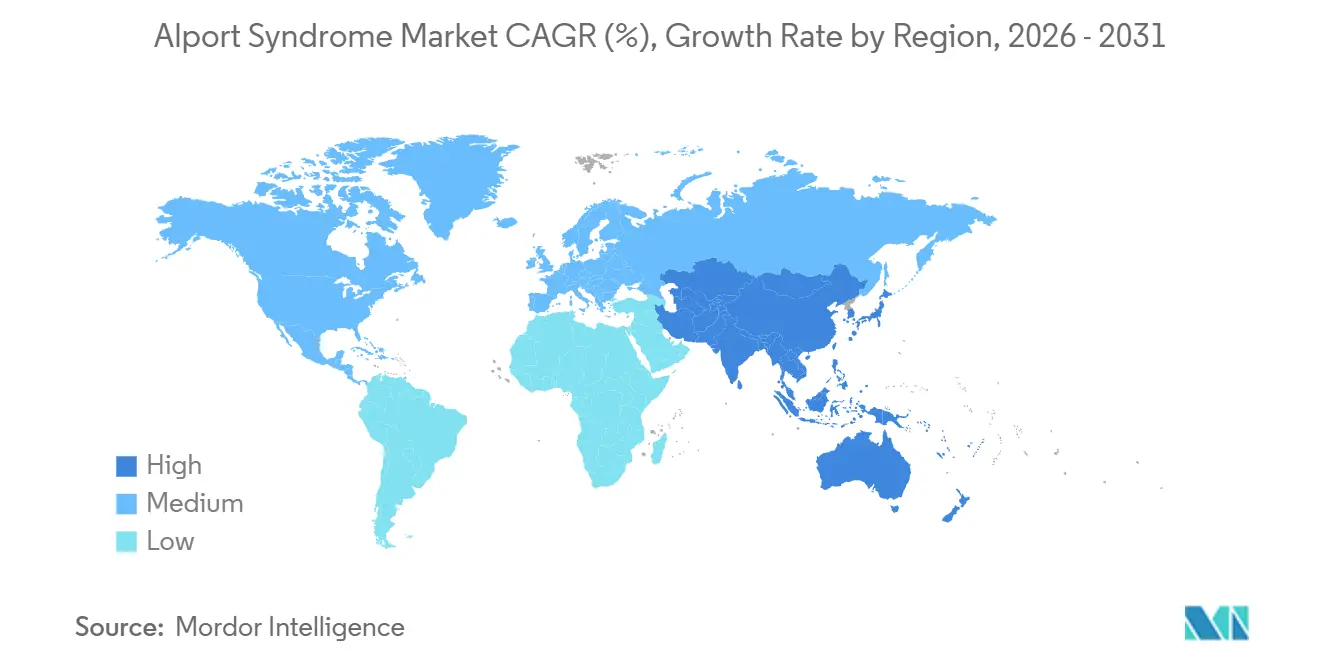
Nach Geografie erfasste Nordamerika im Jahr 2025 einen Anteil von 43,98 %, während der asiatisch-pazifische Raum bis 2031 voraussichtlich mit einem CAGR von 57,12 % wachsen wird.
Hinweis: Die Marktgröße und Prognosezahlen in diesem Bericht werden mithilfe des proprietären Schätzungsrahmens von Mordor Intelligence erstellt und mit den neuesten verfügbaren Daten und Erkenntnissen vom Januar 2026 aktualisiert.
Globale Alport-Syndrom-Markttrends und -Einblicke
Analyse der Treiberwirkung*
| Treiber | (~) % Auswirkung auf die CAGR-Prognose | Geografische Relevanz | Zeithorizont der Auswirkung |
|---|---|---|---|
| Ausweitung der Nutzung genetischer Tests bei vermutetem erblichem Nierenerkrankung | +9.5% | Global | Mittelfristig (2–4 Jahre) |
| Breitere Anwendung früher RAAS-Blockade zur Verzögerung des Fortschreitens | +7.0% | Global | Kurzfristig (≤ 2 Jahre) |
| Zunehmende Studienaktivität in mutationsdefinierten Patientenuntergruppen | +12.0% | Nordamerika und EU | Mittelfristig (2–4 Jahre) |
| Orphan-Drug-Ökonomie unterstützt schnellere Entwicklung für seltene Nierentherapien | +8.5% | Nordamerika und EU | Mittelfristig (2–4 Jahre) |
| Integration von NGS-Panels in der Nephrologie und bei Transplantationsabklärungen | +6.0% | Global, frühe Gewinne im asiatisch-pazifischen Raum | Mittelfristig (2–4 Jahre) |
| Erweitertes familiäres Kaskaden-Screening deckt bisher verborgene Prävalenzfälle auf | +2.5% | Global | Langfristig (≥ 4 Jahre) |
| Quelle: Mordor Intelligence | |||
Zunehmende Studienaktivität in mutationsdefinierten Patientenuntergruppen
Der Alport-Syndrom-Markt verfügt im Jahr 2026 über eine aktivere klinische Pipeline als je zuvor, und neue Programme werden rund um mutationsdefinierte Patientengruppen konzipiert. Bayer initiierte im Dezember 2025 die ASSESS-Phase-IIa-Studie mit BAY 3401016, und das Programm trägt bereits die FDA-Fast-Track- und Orphan-Drug-Designierungen. Eloxx eröffnete im Mai 2026 die EXACT-Phase-2b-Studie für die Aufnahme von Patienten mit bestätigten Nonsense-Mutationen in COL4A3, COL4A4 und COL4A5. Dieses Biomarker-gesteuerte Modell ermöglicht es Sponsoren, genetisch homogenere Kohorten zu untersuchen, und verbessert die Chance, klinisch bedeutsame Effektgrößen in einer kleinen Erkrankungspopulation nachzuweisen. Infolgedessen verfügt der Alport-Syndrom-Markt nun über parallele Zulassungswege, die gleichzeitig voranschreiten, was die Chance auf eine erste zugelassene Therapie wesentlich verbessert.
Orphan-Drug-Ökonomie unterstützt schnellere Entwicklung für seltene Nierentherapien
Die Orphan-Drug-Ökonomie ist zu einem praktischen Entwicklungskatalysator für den Alport-Syndrom-Markt geworden, da sie Kosten senkt und die regulatorische Sichtbarkeit verbessert. Calliditas Therapeutics erhielt die FDA-Orphan-Drug-Designierung für Setanaxib, was die Entwicklungshürden senkte und eine engere regulatorische Einbindung rund um das Programm unterstützte.[2]Calliditas Therapeutics, "Calliditas Therapeutics berichtet Sicherheitsdaten für Setanaxib bei Patienten mit Alport-Syndrom auf der Kidney Week der American Society of Nephrology," PRNewswire, prnewswire.com Das BAY-3401016-Programm von Bayer trägt ebenfalls sowohl Fast-Track- als auch Orphan-Drug-Designierungen, die einen schnelleren Prüfweg unterstützen, sobald eine Einreichung bereit ist. Die Wirtschaftlichkeit hinter diesen Rahmenbedingungen ist nun stark genug, um Partnerschaftsstrukturen anzuziehen, die das Entdeckungsrisiko verteilen und gleichzeitig das kommerzielle Aufwärtspotenzial erhalten, wie die Zusammenarbeit von Bayer und Evotec rund um BAY 3401016 zeigt. Diese Verschiebung macht den Alport-Syndrom-Markt investierbarer als noch vor wenigen Jahren.
Ausweitung der Nutzung genetischer Tests bei vermutetem erblichem Nierenerkrankung
Der Alport-Syndrom-Markt expandiert diagnostisch, da COL4A3-, COL4A4- und COL4A5-Tests nun Teil der Erstlinienabklärung bei ungeklärter persistierender Hämaturie, Proteinurie und fokal-segmentaler Glomerulosklerose unbekannter Ursache sind. NGS-Panels mit breiter Kodierungs- und angrenzender intronischer Abdeckung überschreiten nun eine Sensitivität von 85 % für den Nachweis pathogener Varianten, was die Testleistung stärker macht als bei vielen anderen erblichen Nierenerkrankungen. Die Hauptlücke liegt nun im Überweisungsverhalten, da pädiatrische Patienten mit isolierter mikroskopischer Hämaturie und ohne Familienanamnese häufig übersehen werden, selbst wenn Tests empfohlen werden. Das Schließen dieser Lücke würde den Alport-Syndrom-Markt direkt erweitern, da nicht diagnostizierte Patienten auch die einzige bewährte Frühintervention, die RAAS-Blockade, verpassen, die das Nierenversagen im Median um 18 Jahre verzögern kann.
Breitere Anwendung früher RAAS-Blockade zur Verzögerung des Fortschreitens
Die frühe RAAS-Hemmung bleibt der grundlegende Behandlungstreiber im Alport-Syndrom-Markt, da sie nach wie vor die einzige Intervention mit randomisierter kontrollierter Evidenz in diesem Bereich ist. Die EARLY PRO-TECT Alport-Studie zeigte, dass Ramipril in einer Dosis von 6 mg/m² bei Kindern ab 2 Jahren mit isolierter mikroskopischer Hämaturie oder Mikroalbuminurie sicher und wirksam ist und dass eine frühe Behandlung das Nierenversagen im Median um 18 Jahre verzögern kann.[3]Roser Torra et al., "Diagnose, Management und Behandlung des Alport-Syndroms," Nephrology Dialysis Transplantation, academic.oup.com Die COMBINE-ALPORT-Studie schloss die Erhebung der primären Endpunktdaten im November 2025 ab und testete Dapagliflozin und Spironolacton zusätzlich zur maximalen RAAS-Blockade, was Evidenz für eine mögliche Zusatzrolle bei Erwachsenen mit persistierender Proteinurie liefert. Eine klinisch wichtige Komplikation ist, dass das RAAS-Ansprechen genotyp-stratifiziert ist und Patienten mit trunkierenden COL4A5-Varianten eine geringere Proteinuriereduktion zeigen als Träger von Missense-Varianten. Dieses Muster unterstützt eine frühere Identifizierung von Patienten, die möglicherweise neuartige Wirkstoffe benötigen, anstatt die konventionelle Behandlung allein im Alport-Syndrom-Markt länger fortzuführen.
Analyse der Hemmnisse*
| Hemmnis | (~) % Auswirkung auf die CAGR-Prognose | Geografische Relevanz | Zeithorizont der Auswirkung |
|---|---|---|---|
| Begrenzte Verfügbarkeit krankheitsmodifizierender Behandlungen | -4.5% | Global | Kurzfristig (≤ 2 Jahre) |
| Variables Phänotyp verlangsamt Diagnose und Überweisung | -3.5% | Global, insbesondere Südamerika und MEA | Mittelfristig (2–4 Jahre) |
| Erstattungsreibung bei kostenintensiven Tests und Therapien für seltene Erkrankungen | -3.5% | Nordamerika und EU | Mittelfristig (2–4 Jahre) |
| Fertigungsengpässe bei AAV und fortgeschrittenen Gentherapien | -2.5% | Global | Langfristig (≥ 4 Jahre) |
| Quelle: Mordor Intelligence | |||
Begrenzte Verfügbarkeit krankheitsmodifizierender Behandlungen
Das Fehlen einer zugelassenen krankheitsmodifizierenden Therapie ist nach wie vor die größte strukturelle Begrenzung der kurzfristigen Einnahmen im Alport-Syndrom-Markt. Enyo Pharma berichtete im Januar 2026 Phase-2-Alpestria-1-Daten, die zeigten, dass Vonafexor die eGFR-Trajektorie von einem historischen mittleren Rückgang von -6,4 mL/min/1,73 m²/Jahr auf einen mittleren Anstieg von +4,8 mL/min/1,73 m²/Jahr über 24 Wochen verschob, und 73 % der Teilnehmer hielten den UACR 3 Monate nach Behandlungsende unter dem Ausgangswert.[4]Enyo Pharma, "Enyo Pharma gibt Abschluss und Topline-Daten der klinischen Phase-2-Alpestria-1-Studie beim Alport-Syndrom bekannt," BusinessWire, businesswire.com Selbst mit diesen Ergebnissen war Phase 3 noch für die zweite Hälfte des Jahres 2026 geplant, und eine regulatorische Zulassung war frühestens vor 2028 unwahrscheinlich. Dieses Timing lässt Patienten auf generische unterstützende Versorgung angewiesen und hält die Gesundheitsausgaben in Richtung Dialyse und Transplantation geneigt. Bis eine krankheitsmodifizierende Option den Markt erreicht, wird der Alport-Syndrom-Markt weiterhin ein Missverhältnis zwischen Krankheitslast und therapeutischem Zugang aufweisen.
Erstattungsreibung bei kostenintensiven Tests und Therapien für seltene Erkrankungen
Erstattungsreibung verlangsamt den Alport-Syndrom-Markt, da fortgeschrittene Diagnostik und zukünftige Spezialtherapien einem weitaus schwierigeren Zugangspfad gegenüberstehen als die standardmäßige unterstützende Versorgung. Zukünftige Gentherapie- und Biologika-Kandidaten werden voraussichtlich in Preisbänder eintreten, die bereits in anderen seltenen Erkrankungen mit strenger Kostenträgerbewertung verbunden sind, was bedeutet, dass starke gesundheitsökonomische Evidenz vor einer breiten Akzeptanz erforderlich sein wird. In Europa haben Institutionen wie G-BA, NICE und HAS historisch gesehen mehrjährige Real-World-Evidenz gefordert, bevor sie eine vollständige Erstattung für ultra-seltene Therapien gewähren, was den kommerziellen Zugang weit nach der regulatorischen Zulassung verzögern kann. Diese Zugangsverzögerung bedeutet, dass der Alport-Syndrom-Markt wissenschaftlich weiter wachsen kann, während die kommerzielle Akzeptanz langsamer bleibt als der klinische Bedarf.
*Unsere Prognosen behandeln die Auswirkungen von Treibern und Einschränkungen als richtungsweisend und nicht additiv. Die Wirkungsprognosen berücksichtigen Basiswachstum, Mischungseffekte und Wechselwirkungen zwischen Variablen.
Segmentanalyse
Nach genetischer Mutation: X-chromosomaler Subtyp verankert Einnahmen, Entstehung des autosomal-dominanten Subtyps erweitert die adressierbare Population
Das X-chromosomale Alport-Syndrom hielt im Jahr 2025 75,94 % des Alport-Syndrom-Marktanteils, was sowohl seine höhere klinische Belastung als auch seine stärkere diagnostische Sichtbarkeit in der Praxis widerspiegelt. Unbehandelte Männer mit COL4A5-Mutationen erreichen häufig bis zum Alter von 40 Jahren ein Nierenversagen, was XLAS im Mittelpunkt der nephrologischen Nachsorge, der Nierenintervention und der Krankheitsüberwachungskosten hält. Diese Konzentration schwerer Krankheitslast gab XLAS eine überproportionale Rolle bei der aktuellen Einnahmenverteilung im Alport-Syndrom-Markt. Innerhalb von XLAS beginnt die genotyp-informierte RAAS-Titration die Behandlungsintensität und den Zeitpunkt des Übergangs für Patienten zu beeinflussen, die am wenigsten wahrscheinlich auf die Standardversorgung ansprechen.
Das autosomal-dominante Alport-Syndrom wird voraussichtlich von 2026 bis 2031 mit einem CAGR von 52,87 % wachsen, was es zum am schnellsten wachsenden Mutationssegment macht. Populationsgenomische Datensätze zeigen, dass ADAS weit häufiger ist als ältere klinische Fallserien vermuten ließen, was bedeutet, dass viele betroffene Personen zuvor übersehen wurden, da die Präsentation milder und variabler ist. Das Kaskaden-Screening im Rahmen des ERKNet-Rahmens bringt nun heterozygote Verwandte in den diagnostizierten Pool, was die adressierbare Basis des Alport-Syndrom-Marktes ohne jede Änderung der Inzidenz erweitert. Autosomal-rezessive und digene Erkrankungen bleiben in der Anzahl kleiner, aber ihre schweren oder neu erkannten Präsentationen unterstützen mehr Tests und eine stabilere Klassifizierung, da Multi-Gen-Panels engere Abklärungen ersetzen.
der



Nach Behandlungsart: Unterstützende Pharmakotherapie dominiert, Wachstum der Nierenersatztherapie spiegelt anhaltenden ungedeckten Bedarf wider
Die unterstützende pharmakologische Therapie machte im Jahr 2025 65,11 % der Alport-Syndrom-Marktgröße aus, was sie zur zentralen Einnahmenbasis über alle Behandlungskategorien hinweg macht. Ramipril bleibt die am besten untersuchte Option, und die EARLY PRO-TECT-Evidenz zeigte, dass eine frühe RAAS-Behandlung das Nierenversagen bei Kindern sicher um einen Median von 18 Jahren verzögern kann. Diese Evidenz hält die unterstützende Versorgung als Standardeinstiegspunkt für die meisten bestätigten Patienten im Alport-Syndrom-Markt. Die Zusatzanwendung von SGLT2-Inhibitoren findet auch bei Erwachsenen mit persistierender Albuminurie trotz maximaler RAAS-Dosierung Eingang in die Praxis, was die kommerzielle Rolle der unterstützenden Therapie erweitert.
Die Nierenersatztherapie wird bis 2031 voraussichtlich mit einem CAGR von 55,34 % wachsen, dem schnellsten Tempo unter den Behandlungssegmenten. Dieser Anstieg spiegelt eine größere diagnostizierte Population wider, die das Endstadium der Erkrankung erreicht, bevor eine zugelassene krankheitsmodifizierende Therapie kommerziell verfügbar wird. Aufkommende Therapien wie Vonafexor, Setanaxib, BAY 3401016 und Exaluren tragen derzeit begrenzte Einnahmen bei, tragen aber den größten Teil des zukünftigen Aufwärtspotenzials für den Alport-Syndrom-Markt. Über diese Pipeline hinaus legt frühe mRNA-Lipid-Nanopartikel-Arbeit in XLAS-Mausmodellen nahe, dass kurative Ansätze die Alport-Syndrom-Branche letztendlich über das unterstützende Krankheitsmanagement hinausbewegen könnten, obwohl diese Möglichkeit präklinisch bleibt.
Nach Vertriebskanal: Krankenhausapotheken führen, Spezialapotheken sind für die Markteinführungsbereitschaft positioniert
Krankenhausapotheken machten im Jahr 2025 53,18 % der Alport-Syndrom-Marktgröße aus, was die spezialistengeführte Natur der Verschreibung und Abgabe bei seltenen Nierenerkrankungen widerspiegelt. Tertiärzentren und Transplantationsprogramme verwalten nach wie vor den größten Teil der Patientenüberwachung, der standardmäßigen unterstützenden Therapie und des Zugangs zu Prüfmedikamenten im Alport-Syndrom-Markt. Diese Struktur hält Krankenhaussysteme tief in den Versorgungspfad eingebettet, da sie auch die kontrollierte Lagerung und die Unterstützung der Lieferkette für die klinische Studienversorgung bereitstellen. Der Kanal profitiert daher sowohl von der aktuellen Versorgungsnachfrage als auch von den betrieblichen Anforderungen der aktiven Pipeline.
Spezialapotheken werden voraussichtlich von 2026 bis 2031 mit einem CAGR von 53,17 % wachsen, was sie zum am schnellsten wachsenden Vertriebskanal macht. Ihre Attraktivität ergibt sich aus Kühlkettenlogistik, Unterstützung bei der Vorabgenehmigung, Patientenservices und der Koordination mit Herstellerhilfsprogrammen, die Krankenhausapotheken nicht immer in kommerziellem Maßstab bereitstellen. Einzelhandelsapotheken versorgen weiterhin die ACE-Hemmer- und ARB-Erhaltungstherapie, während Online-Apotheken als Komfortschicht für die Wiederholungsabgabe in unterversorgten Regionen entstehen. Da Biologika und andere Premium-Therapien der Markteinführung näher rücken, wird sich der Vertrieb im Alport-Syndrom-Markt wahrscheinlich in Richtung Spezialapotheken-Infrastruktur verlagern.
Geografische Analyse
Nordamerika hielt im Jahr 2025 43,98 % des Alport-Syndrom-Marktanteils und war damit der größte regionale Beitragszahler. Die Region profitiert von dichten nephrologischen Spezialistennetzwerken, akademischen Zentren für seltene Nierenerkrankungen und einem Orphan-Drug-Rahmen, der das Entwicklungsrisiko für Sponsoren senkt. Die Alport Syndrome Foundation führte im Dezember 2025 auch direkte Gespräche mit der FDA über Studienendpunkte und patientenorientierte Arzneimittelentwicklungsprioritäten, was ein klareres Studiendesign für den Alport-Syndrom-Markt unterstützt. Die Vereinigten Staaten bleiben das wichtigste Einnahmenzentrum, während Kanada durch provinzielle Abdeckung Unterstützung hinzufügt, die zunehmend erbliche Nephropathie-Panels in der pädiatrischen Versorgung einschließt.
Europa blieb die zweite wichtige regionale Basis für den Alport-Syndrom-Markt, unterstützt durch das ERKNet-Referenznetzwerk in Deutschland, Frankreich, den Niederlanden, Spanien und Italien. Diese Länder haben bereits als führende Studienstandorte für Vonafexor und Setanaxib gedient, was der Region praktische Erfahrung in der Durchführung seltener Nierenstudien verleiht. Deutschlands DOUBLE PRO-TECT Alport Phase-3-Studie rekrutiert Jugendliche und junge Erwachsene für die Dapagliflozin-Bewertung, und dieser unabhängige Evidenzstrom könnte zukünftige Leitlinienaktualisierungen und die Akzeptanz durch Kostenträger in der gesamten Region beeinflussen.[5]"DOUBLE PRO-TECT Alport, multizentrische, randomisierte, placebokontrollierte Studie mit Dapagliflozin bei Jugendlichen und jungen Erwachsenen mit Alport-Syndrom," Orphanet, orpha.net Europa verbindet daher eine starke klinische Beteiligung am Alport-Syndrom-Markt mit strengeren Evidenzanforderungen für die Erstattung.
Der asiatisch-pazifische Raum wird voraussichtlich von 2026 bis 2031 mit einem CAGR von 57,12 % wachsen, was ihn zur am schnellsten expandierenden Geografie im Alport-Syndrom-Markt macht. Japans obligatorisches Urinanalyseprogramm ist zu einem Früherkennungspfad für pädiatrische Fälle geworden, und das JP-ALPS-Register gibt der Region eine stärkere molekulare und klinische Referenzbasis. China erweitert auch die Testkapazitäten in Tertiärkrankenhäusern, und Arbeiten aus dem Südwesten Chinas identifizierten neuartige COL4A3-, COL4A4- und COL4A5-Varianten zusammen mit digenen Vererbungsmustern, die auf eine noch unvollständige diagnostische Karte hinweisen.

Wettbewerbslandschaft
Der Alport-Syndrom-Markt hat eine gespaltene Wettbewerbsstruktur, mit einem mäßig fragmentierten Diagnostiksegment und einem Therapiesegment, das noch keinen zugelassenen Marktführer hat. In der Diagnostik stellt Illumina die zentrale Sequenzierungsinfrastruktur bereit, während Invitae, Natera, CENTOGENE, Eurofins, Quest Diagnostics und andere Anbieter von erblichen Nephropathie-Panels auf der Dienstleistungsebene konkurrieren. Der Wettbewerb in diesem Teil des Alport-Syndrom-Marktes hängt weniger vom Sequenzierungszugang allein ab und mehr von der Panel-Breite, der Bearbeitungszeit, den Kostenträgerverträgen und der Qualität der Variantenklassifizierung.
In der Therapeutik entwickeln Enyo Pharma, Calliditas Therapeutics, Bayer AG und Eloxx Pharmaceuticals unterschiedliche Mechanismen, was den Alport-Syndrom-Markt offen statt konsolidiert hält. Vonafexor zielt auf FXR ab, Setanaxib zielt auf fibrose-assoziierte NOX-Wege ab, BAY 3401016 zielt auf Semaphorin 3A ab, und Exaluren wird auf Nonsense-Mutations-Readthrough getestet. Diese mechanistische Vielfalt legt nahe, dass die zukünftige Versorgung möglicherweise nicht auf einem universellen Produkt zentriert sein wird, insbesondere wenn das Ansprechen weiterhin je nach Genotyp und Krankheitsstadium variiert. Bayer stärkte seine Position, als es BAY 3401016 im Dezember 2025 in Phase IIa überführte, nachdem die Zusammenarbeit von Bayer und Evotec das Programm hervorgebracht hatte. Eloxx trieb das Feld auch voran, indem es im Mai 2026 die EXACT-Phase-2b-Studie für mutationsselektierte Patienten eröffnete, was zeigt, wie die gezielte Entwicklung zu einem Kernmuster im Alport-Syndrom-Markt wird.
Strategische Transaktionsaktivitäten prägen das Feld ebenfalls, und die Übernahme von Renalys durch Chugai Pharmaceutical im vierten Quartal 2025 verlängerte den Weg von Sparsentan zur japanischen Überprüfung für das Alport-Syndrom und löste bei Abschluss eine Zahlung von 10,2 Millionen USD an Travere Therapeutics aus. Illuminas fortlaufende NovaSeq-Investitionen und Populationsgenomik-Partnerschaften unterstützen die vorgelagerte Testkapazität, von der das Screening auf seltene erbliche Nephropathien abhängt. Weißer Raum verbleibt in pädiatrischen Formulierungen, Begleitdiagnostika und Gentherapiekonstrukten, sodass der Alport-Syndrom-Markt noch Raum für neue Marktteilnehmer bietet, auch wenn mehrere namentlich genannte Entwickler bereits vorangeschritten sind. Diese Kombination aus aktiven Entwicklern, offenem First-Mover-Status und unvollendeter diagnostischer Standardisierung hält den Alport-Syndrom-Markt dynamisch, aber noch weit von einer Konsolidierung entfernt.
Alport-Syndrom-Branchenführer
Bayer AG
Calliditas Therapeutics AB
Chinook Therapeutics, Inc.
Novartis AG
Travere Therapeutics, Inc.
- *Haftungsausschluss: Hauptakteure in keiner bestimmten Reihenfolge sortiert

Jüngste Branchenentwicklungen
- Mai 2026: Eloxx Pharmaceuticals eröffnete die Aufnahme für die EXACT-Phase-2b-Studie (NCT07523581), eine randomisierte, doppelblinde, placebokontrollierte Studie mit verzögertem Start, die Exaluren bei Patienten mit X-chromosomalem oder autosomal-rezessivem Alport-Syndrom mit bestätigten COL4A3/4/5-Nonsense-Mutationen bewertet, mit dem Ziel, 24 Patienten ab 12 Jahren an Standorten in den USA und im Vereinigten Königreich einzuschließen. Dies ist das erste mutationsklassenspezifische Nonsense-Readthrough-Programm, das Phase 2b beim Alport-Syndrom erreicht, mit erwartetem primärem Abschluss im Juni 2027 und vollständigem Studienabschluss im Dezember 2027.
- Januar 2026: Enyo Pharma veröffentlichte Topline-Phase-2-Alpestria-1-Daten, die zeigten, dass Vonafexor die eGFR-Trajektorie von einem historischen mittleren Rückgang von -6,4 mL/min/1,73 m²/Jahr auf einen mittleren Anstieg von +4,8 mL/min/1,73 m²/Jahr über 24 Wochen Behandlung bei 26 Hochrisiko-Alport-Syndrom-Patienten unter Standardversorgung umkehrte, wobei 73 % der Patienten die UACR-Reduktion unter den Ausgangswert drei Monate nach Behandlungsende aufrechterhielten. Ein End-of-Phase-2-FDA-Treffen ist für das zweite Quartal 2026 geplant, und die Einleitung von Phase 3 ist für die zweite Hälfte des Jahres 2026 angestrebt.
- Dezember 2025: Bayer AG initiierte die ASSESS-Phase-IIa-Studie (NCT07211685) mit BAY 3401016, einem investigativen Anti-Semaphorin-3A-monoklonalen Antikörper, der aus der Bayer-Evotec-Multi-Target-Nierenforschungskooperation stammt, bei erwachsenen XLAS- oder ARAS-Patienten mit erhöhter Albuminurie. Das Programm trägt FDA-Fast-Track- und Orphan-Drug-Designierungen und etabliert Bayer als das größte Pharmaunternehmen mit einem aktiven krankheitsmodifizierenden Alport-Syndrom-Therapieprogramm.
- November 2025: Calliditas Therapeutics, ein Unternehmen von Asahi Kasei, präsentierte Phase-2a-Sicherheits- und sekundäre Wirksamkeitsergebnisse für Setanaxib auf der ASN Kidney Week High-Impact Clinical Trials-Sitzung in Houston. Die Studie (NCT06274489) schloss 20 Patienten im Alter von 12–40 Jahren mit genetisch bestätigtem Alport-Syndrom ein und erfüllte ihre primären Sicherheitsendpunkte, während Setanaxib eine mittlere UPCR-Reduktion von 15 % nach 24 Wochen und eine mittlere UPCR-Reduktion von 27 % 4 Wochen nach der Dosierung gegenüber Placebo erzielte, was auf einen anhaltenden antifibrotischen Effekt hindeutet.


Globaler Alport-Syndrom-Marktbericht Umfang
Der Alport-Syndrom-Markt umfasst Therapeutika, Diagnosetechnologien und unterstützende Versorgungslösungen, die auf die Diagnose und das Management des Alport-Syndroms abzielen, einer seltenen erblichen Nierenerkrankung, die durch Mutationen in Kollagen-Typ-IV-Genen verursacht wird. Der Markt wird durch die zunehmende Nutzung genetischer Tests, ein wachsendes Bewusstsein für seltene Nierenerkrankungen, Fortschritte in der nephrologischen Versorgung und die Entwicklung gezielter krankheitsmodifizierender Therapien angetrieben. Fortgesetzte Investitionen in die Orphan-Drug-Forschung, Präzisionsmedizin und Frühdiagnose werden voraussichtlich die Patientenergebnisse verbessern und das Marktwachstum über den Prognosezeitraum hinweg unterstützen.
Der Alport-Syndrom-Markt ist nach genetischer Mutation, Behandlungsart, Vertriebskanal und Geografie segmentiert. Nach genetischer Mutation ist er weiter unterteilt in X-chromosomales Alport-Syndrom, autosomal-dominantes Alport-Syndrom, autosomal-rezessives Alport-Syndrom und digenes Alport-Syndrom. Nach Behandlungsart ist er segmentiert in unterstützende pharmakologische Therapie, Nierenersatztherapie und aufkommende krankheitsmodifizierende Therapien. Nach Vertriebskanal ist der Markt segmentiert in Krankenhausapotheken, Einzelhandelsapotheken, Spezialapotheken und Online-Apotheken. Das Geografiesegment ist weiter unterteilt in Nordamerika, Europa, den asiatisch-pazifischen Raum und den Rest der Welt. Der Bericht deckt auch die geschätzten Marktgrößen und Trends für 13 Länder in den wichtigsten Regionen weltweit ab. Der Bericht bietet die Marktgröße und Prognosen in Wert (USD) für die oben genannten Segmente.
| X-chromosomales Alport-Syndrom |
| Autosomal-dominantes Alport-Syndrom |
| Autosomal-rezessives Alport-Syndrom |
| Digenes Alport-Syndrom |
| Unterstützende pharmakologische Therapie |
| Nierenersatztherapie |
| Aufkommende krankheitsmodifizierende Therapien |
| Krankenhausapotheken |
| Einzelhandelsapotheken |
| Spezialapotheken |
| Online-Apotheken |
| Nordamerika | Vereinigte Staaten |
| Kanada | |
| Mexiko | |
| Europa | Deutschland |
| Vereinigtes Königreich | |
| Frankreich | |
| Italien | |
| Spanien | |
| Übriges Europa | |
| Asiatisch-pazifischer Raum | China |
| Japan | |
| Indien | |
| Australien | |
| Südkorea | |
| Übriger asiatisch-pazifischer Raum | |
| Rest der Welt |
| Nach genetischer Mutation | X-chromosomales Alport-Syndrom | |
| Autosomal-dominantes Alport-Syndrom | ||
| Autosomal-rezessives Alport-Syndrom | ||
| Digenes Alport-Syndrom | ||
| Nach Behandlungsart | Unterstützende pharmakologische Therapie | |
| Nierenersatztherapie | ||
| Aufkommende krankheitsmodifizierende Therapien | ||
| Nach Vertriebskanal | Krankenhausapotheken | |
| Einzelhandelsapotheken | ||
| Spezialapotheken | ||
| Online-Apotheken | ||
| Nach Geografie | Nordamerika | Vereinigte Staaten |
| Kanada | ||
| Mexiko | ||
| Europa | Deutschland | |
| Vereinigtes Königreich | ||
| Frankreich | ||
| Italien | ||
| Spanien | ||
| Übriges Europa | ||
| Asiatisch-pazifischer Raum | China | |
| Japan | ||
| Indien | ||
| Australien | ||
| Südkorea | ||
| Übriger asiatisch-pazifischer Raum | ||
| Rest der Welt | ||


Im Bericht beantwortete Schlüsselfragen
Welchen prognostizierten Wert wird der Alport-Syndrom-Bereich bis 2031 erreichen?
Der Alport-Syndrom-Markt wird voraussichtlich bis 2031 530,23 Millionen USD erreichen, ausgehend von 52,65 Millionen USD im Jahr 2025, mit einem CAGR von 46,95 % über den Zeitraum 2026–2031.
Warum beschleunigt sich das Wachstum in diesem Bereich so schnell?
Das Wachstum wird durch aktive Phase-2- und Phase-2b-Pipelines, breitere genetische Tests, frühere Diagnose durch Kaskaden-Screening und die fortgesetzte Anwendung RAAS-basierter unterstützender Behandlung angetrieben.
Welcher genetische Subtyp trägt derzeit am meisten zum Umsatz bei?
Das X-chromosomale Alport-Syndrom führte im Jahr 2025 mit einem Anteil von 75,94 %, da seine klinische Schwere und höhere Versorgungsintensität mehr spezialisierte Nachsorge und Behandlungsausgaben generieren.
Welche Behandlungskategorie wächst bis 2031 am schnellsten?
Die Nierenersatztherapie wird voraussichtlich mit einem CAGR von 55,34 % wachsen, was das anhaltende Fortschreiten zur Endstadiumerkrankung widerspiegelt, bevor zugelassene krankheitsmodifizierende Therapien verfügbar werden.
Welche Region wird voraussichtlich am schnellsten expandieren?
Der asiatisch-pazifische Raum wird voraussichtlich bis 2031 mit einem CAGR von 57,12 % wachsen, unterstützt durch Japans Screening-Pfad und Chinas expandierende genetische Testkapazität.
Was ist die größte kurzfristige kommerzielle Hürde für Entwickler?
Die Haupthürde ist das Fehlen einer zugelassenen krankheitsmodifizierenden Therapie, gefolgt von Kostenträgerreibung bei fortgeschrittenen Tests und zukünftigen kostenintensiven Behandlungen für seltene Erkrankungen.
Seite zuletzt aktualisiert am: