Marktgröße und Marktanteil im Bereich Medical Device Regulatory Affairs
Marktübersicht
| Studienzeitraum | 2020 - 2031 |
|---|---|
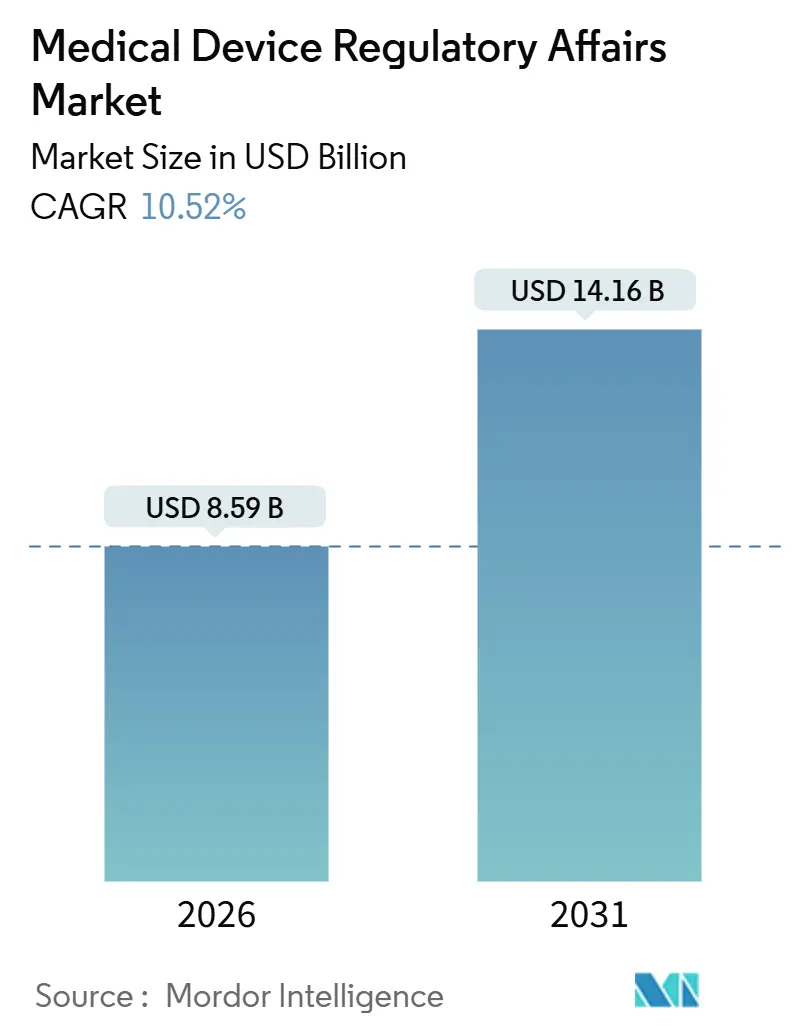
| Marktgröße (2026) | 8.59 Milliarden US-Dollar |
| Marktgröße (2031) | 14.16 Milliarden US-Dollar |
| Wachstumsrate (2026 - 2031) | 10.52% CAGR |
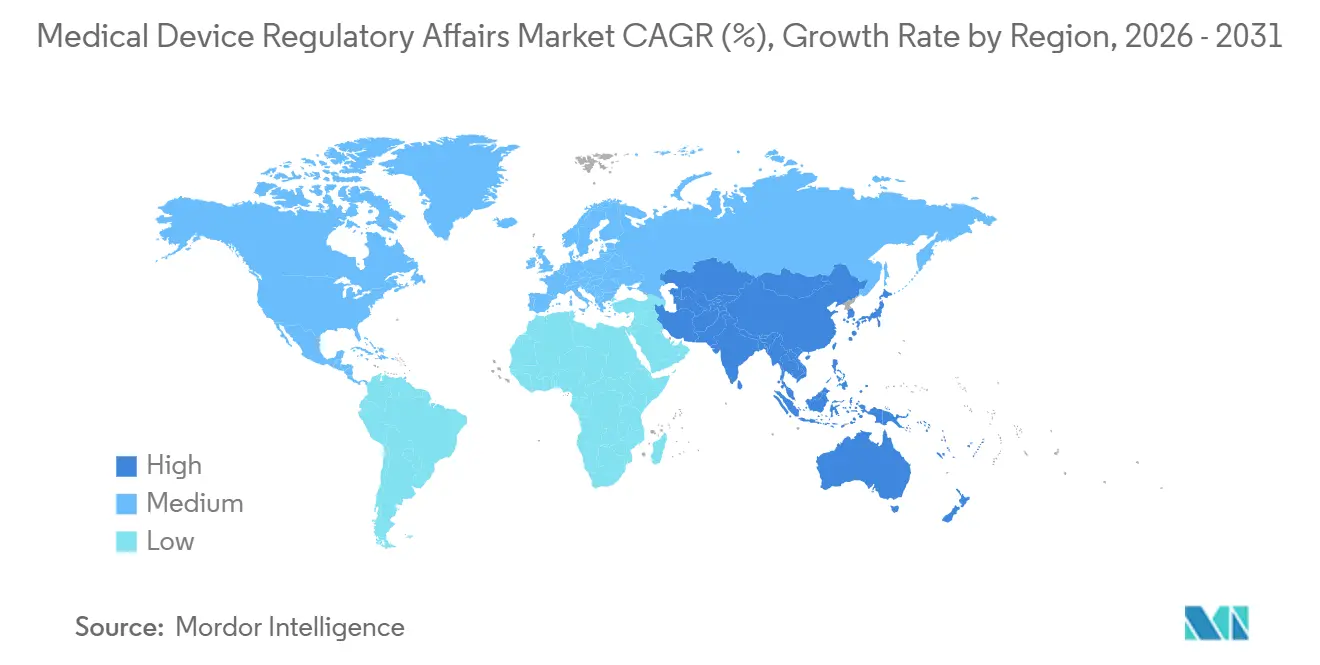
| Schnellstwachsender Markt | Asien-Pazifik |
| Größter Markt | Nordamerika |
| Marktkonzentration | Mittel |
Hauptakteure *Haftungsausschluss: Hauptakteure in keiner bestimmten Reihenfolge sortiert Bild © Mordor Intelligence. Wiederverwendung erfordert Namensnennung gemäß CC BY 4.0. | |
Marktanalyse für Medical Device Regulatory Affairs von Mordor Intelligence
Die Marktgröße für Medical Device Regulatory Affairs wird im Jahr 2026 auf 8,59 Milliarden USD geschätzt und soll bis 2031 14,16 Milliarden USD erreichen, bei einer CAGR von 10,52 % während des Prognosezeitraums (2026–2031).
Anhaltende regulatorische Verschärfungen, steigende Einreichungsvolumina und der Wettlauf um die Kommerzialisierung digitaler Gesundheitsinnovationen veranlassen Hersteller dazu, in allen wichtigen Regionen spezialisierte Beratung zu suchen. Outsourcing bleibt das vorherrschende Betriebsmodell, da Beratungsverträge mit variablen Kosten die Budgets vor den zyklischen Arbeitsspitzen schützen, die durch neue Vorschriften wie das FDA-Cybersicherheitsmandat vom März 2024 ausgelöst werden. Cloud-native Plattformen für das regulatorische Informationsmanagement (RIM) gestalten die täglichen Arbeitsabläufe neu, indem sie die Dokumentenzusammenstellung und Echtzeit-Lückenprüfungen automatisieren. Gleichzeitig halten demografische Entwicklungen in alternden Volkswirtschaften die Gerätepipelines aufrecht, was wiederum die Liste der erforderlichen regulatorischen Einreichungen verlängert. Die Wettbewerbsintensität ist moderat, doch Erstmovervorteile entstehen für Anbieter, die US-amerikanische, EU- und asiatisch-pazifische Einreichungen synchronisieren können, ohne Geschwindigkeit oder Qualität zu beeinträchtigen.
Wichtigste Erkenntnisse des Berichts
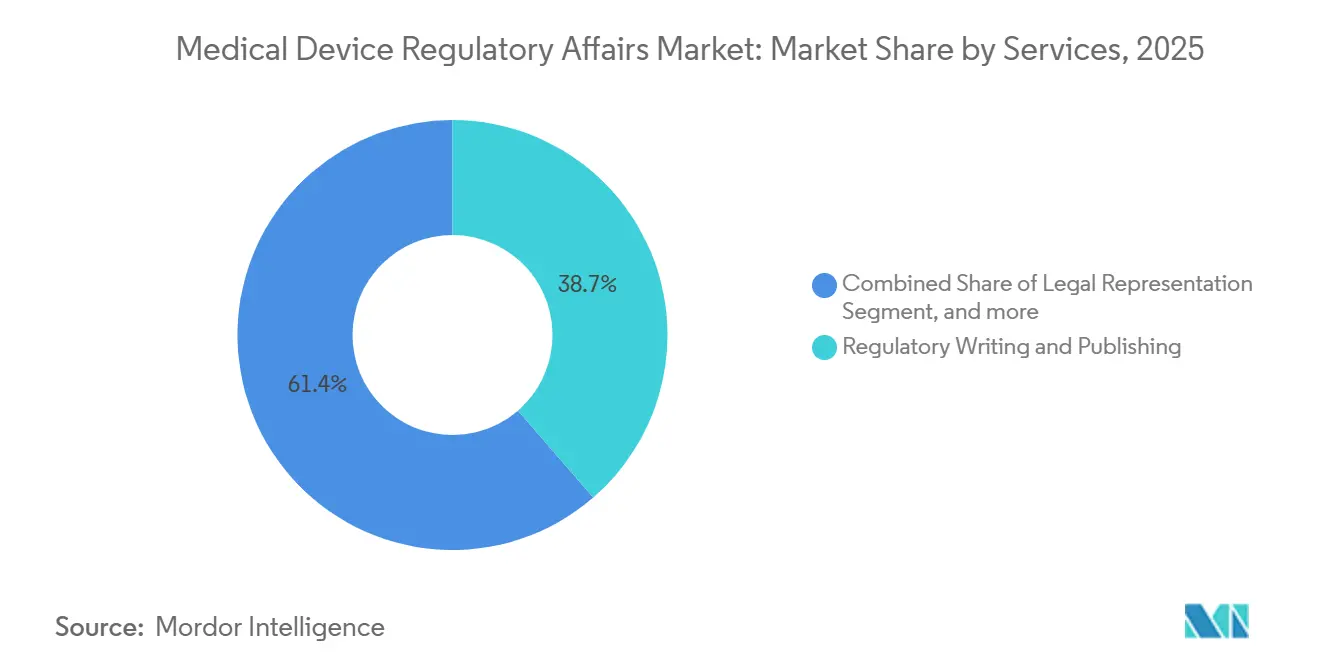
- Nach Dienstleistungsanbieter entfielen auf ausgelagerte Spezialisten 58,65 % des Marktanteils für Medical Device Regulatory Affairs im Jahr 2025. Produktregistrierung und klinische Studienanträge verzeichneten mit 12,76 % die höchste CAGR aller Dienstleistungskategorien bis 2031 in Bezug auf die Marktgröße für Medical Device Regulatory Affairs.
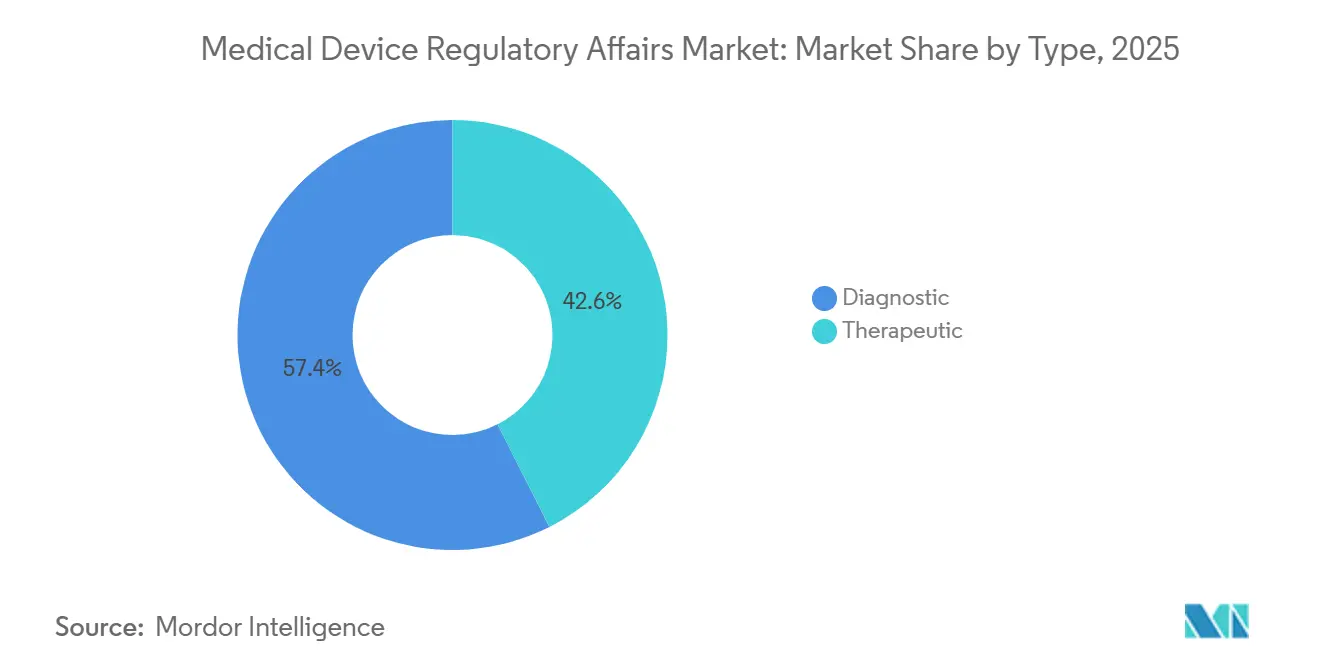
- Nach Typ entfielen auf therapeutische Geräte 42,56 % des Umsatzes im Jahr 2025, während Diagnoseplattformen mit einer marktführenden CAGR von 12,87 % bis 2031 wachsen.
- Nach Dienstleistungsanbieter entfielen auf ausgelagerte Anbieter 58,65 % des Umsatzes im Jahr 2025, während interne Anbieter mit einer marktführenden CAGR von 13,54 % bis 2031 wachsen.
- Nach Geografie führte Nordamerika die Ausgaben mit 42,48 % des globalen Umsatzes im Jahr 2025 an; Asien-Pazifik wächst am schnellsten mit einer CAGR von 11,54 %.
Hinweis: Die Marktgröße und Prognosezahlen in diesem Bericht werden mithilfe des proprietären Schätzungsrahmens von Mordor Intelligence erstellt und mit den neuesten verfügbaren Daten und Erkenntnissen vom Januar 2026 aktualisiert.
Globale Markttrends und Erkenntnisse im Bereich Medical Device Regulatory Affairs
Analyse der Treiberwirkung*
| Treiber | (~) % Auswirkung auf die CAGR-Prognose | Geografische Relevanz | Zeithorizont der Auswirkung |
|---|---|---|---|
| Steigende globale Nachfrage nach innovativen Medizinprodukten | +2.3% | Global, Konzentration in Nordamerika und EU | Mittelfristig (2–4 Jahre) |
| Strenge regulatorische Rahmenbedingungen zur Verbesserung der Patientensicherheit | +2.1% | Nordamerika, EU, Japan | Kurzfristig (≤ 2 Jahre) |
| Wachsende Belastung durch chronische Krankheiten und alternde Bevölkerungen | +1.8% | Global, Höhepunkt in Asien-Pazifik und Nordamerika | Langfristig (≥ 4 Jahre) |
| Expansion von Geräteherstellern in Schwellenmärkten | +1.6% | Asien-Pazifik als Kern, Ausstrahlungseffekte auf Naher Osten und Afrika sowie Südamerika | Mittelfristig (2–4 Jahre) |
| Verbreitung von KI-gestützten Plattformen für regulatorische Informationen | +1.4% | Nordamerika, EU, fortgeschrittenes Asien-Pazifik | Kurzfristig (≤ 2 Jahre) |
| Anforderungen an die Post-Merger-Integration für harmonisierte Compliance | +1.3% | Global, Konzentration in Nordamerika und EU | Kurzfristig (≤ 2 Jahre) |
| Quelle: Mordor Intelligence | |||
Steigende globale Nachfrage nach innovativen Medizinprodukten
Der Anstieg softwaregestützter Diagnostika und minimal-invasiver Therapeutika verkürzt Produktlebenszyklen und vervielfacht Einreichungsereignisse. Die FDA erteilte im Jahr 2025 89 De-Novo-Zulassungen für neuartige Diagnosealgorithmen, ein Anstieg von 102 % gegenüber 2023, und die Einreichungen für Software als Medizinprodukt stiegen um 34 % im Jahresvergleich. Die Koordinierungsgruppe für Medizinprodukte in Europa erweiterte im Februar 2025 die Validierungsleitlinien, die eine vollständige Lebenszyklusdokumentation erfordern und die durchschnittliche Auftragsdauer um sechs Monate verlängern. Hersteller stützen sich daher auf Beratungsunternehmen, um US-amerikanische, EU- und asiatisch-pazifische Dossiers gleichzeitig zu synchronisieren und die Erstmarktpositionierung zu sichern.
Wachsende Belastung durch chronische Krankheiten und alternde Bevölkerungen
Nichtübertragbare Krankheiten waren laut der Weltgesundheitsorganisation im Jahr 2024 für 74 % der weltweiten Todesfälle verantwortlich. Japans Anteil der Bürger ab 65 Jahren erreichte 2024 29,1 % und trieb einen Anstieg der Geräteanträge für die Altenpflege um 15 % an. Die Leitlinien der FDA zu Real-World-Evidence aus dem Jahr 2025 ermöglichen die Integration von Daten aus elektronischen Patientenakten in Einreichungen und erweitern den Aufwand für die Evidenzgenerierung, den Beratungsteams koordinieren müssen.
Expansion von Geräteherstellern in Schwellenmärkten
Beschleunigte Zulassungswege verkürzen die Genehmigungsfristen in Asien-Pazifik und Lateinamerika. Chinas Prioritätsprüfungskanal halbiert die Bewertungszeit für Klasse-III-Geräte auf 9 Monate. Indiens Regelanpassung vom August 2024 reduzierte die Anforderungen an ausländische Dossiers um 30 %, und Brasilien senkte im März 2025 die Gebühren für Kleinunternehmen um 30 %, während es bestimmte FDA-Zulassungen für die gegenseitige Anerkennung akzeptierte. Diese Reformen treiben die ausgelagerte Nachfrage an, wo lokale regulatorische Fachkenntnisse begrenzt sind.
Anforderungen an die Post-Merger-Integration für harmonisierte Compliance
Die Konsolidierung in der Medizinprodukteindustrie schafft multinationale Portfolios, die einheitliche Qualitätssysteme erfordern. Medtronics Kauf von EOFlow für 738 Millionen USD kombinierte Dossiers, die durch unterschiedliche Vorschriften geregelt werden, und erforderte ein 14-monatiges Harmonisierungsprojekt, das von einem gemischten internen und ausgelagerten Team geleitet wurde. Das Internationale Forum der Medizinproduktebehörden skizzierte im November 2025 eine gemeinsame Vorlage für die Meldung unerwünschter Ereignisse und veranlasste Erwerber, vorab in Datenarchitekturen zu investieren, die eine unternehmensweite Überwachung ermöglichen[1]Internationales Forum der Medizinproduktebehörden, "Harmonisierte Meldung unerwünschter Ereignisse," imdrf.org.
Analyse der Hemmnisse*
| Hemmnis | (~) % Auswirkung auf die CAGR-Prognose | Geografische Relevanz | Zeithorizont der Auswirkung |
|---|---|---|---|
| Komplexe und divergierende globale regulatorische Anforderungen | -1.2% | Global, akut in aufstrebendem Asien-Pazifik und Naher Osten und Afrika | Mittelfristig (2–4 Jahre) |
| Hohe Kosten der regulatorischen Compliance | -0.9% | Global, Konzentration in Nordamerika und EU | Kurzfristig (≤ 2 Jahre) |
| Mangel an erfahrenem Fachpersonal im Bereich Regulatory Affairs | -0.7% | Global, Höhepunkt in Nordamerika und EU | Langfristig (≥ 4 Jahre) |
| Cybersicherheitsmandate erhöhen den Dokumentationsaufwand | -0.6% | Nordamerika, EU, fortgeschrittenes Asien-Pazifik | Kurzfristig (≤ 2 Jahre) |
| Quelle: Mordor Intelligence | |||
Komplexe und divergierende globale regulatorische Anforderungen
Das Internationale Forum der Medizinproduktebehörden zählte im Jahr 2024 23 einzigartige Klassifizierungsschemata, die häufig doppelte Tests erfordern, wenn ein Gerät über Grenzen hinweg die Kategorie wechselt. Indiens risikobasierter Rahmen weicht sowohl von den FDA- als auch von den EU-Regeln ab, während Brasilien portugiesischsprachige Kennzeichnung und lokale klinische Daten verlangt, was bis zu sechs Monate zusätzliche Arbeit bedeutet[2]Indische Zentralbehörde für Arzneimittelkontrolle, "Änderung der Medizinprodukteverordnung 2024," cdsco.gov.in. Eine solche Fragmentierung untergräbt die wirtschaftliche Grundlage globaler Markteinführungsstrategien und dämpft die Beratungseinnahmen in weniger priorisierten Regionen.
Mangel an erfahrenem Fachpersonal im Bereich Regulatory Affairs
Die weltweite Mitgliedschaft in der Gesellschaft für Fachleute im Bereich Regulatory Affairs erreichte im Jahr 2025 nach einem Wachstum von 3,1 % nur 18.500 Mitglieder, weit unter der Ausweitung des Arbeitsvolumens um 10,52 %[3]Gesellschaft für Fachleute im Bereich Regulatory Affairs, "Mitgliederstatistik 2025," raps.org. Stellen für leitende Manager bleiben in Nordamerika durchschnittlich 89 Tage unbesetzt und treiben die Arbeitskosten bei Unternehmen wie IQVIA um 6,8 % in die Höhe. Knappheit erhöht die Preise, begrenzt aber auch die Anzahl gleichzeitiger Projekte, die Beratungsunternehmen annehmen können.
*Unsere Prognosen behandeln die Auswirkungen von Treibern und Einschränkungen als richtungsweisend und nicht additiv. Die Wirkungsprognosen berücksichtigen Basiswachstum, Mischungseffekte und Wechselwirkungen zwischen Variablen.
Segmentanalyse
Nach Dienstleistungen: Dokumentationskomplexität treibt die Nachfrage nach Verfassungsleistungen an
Regulatorisches Schreiben und Publizieren erfasste 38,65 % des Umsatzes im Jahr 2025 und spiegelt die dokumentenintensiven Verpflichtungen wider, die durch Cybersicherheitsanhänge und Softwarelebenszykluskontrollen entstehen. Dienstleistungen für Produktregistrierung und klinische Studienanträge, die mit einer CAGR von 12,76 % wachsen, sind der wichtigste Wachstumsmotor, da Reformen in China und Indien die Prüfzeiten halbiert oder reduziert haben und Hersteller dazu verleiten, früher und in mehr Ländern einzureichen. Regulierungsberatung, die Strategieplanung und Lückenanalysen umfasst, verzeichnete eine solide Expansion, da Kunden auf EUDAMED umstellten und US-amerikanische Cybersicherheitsdossiers aktualisierten. Rechtliche Vertretung stieg zusammen mit einem Anstieg von 19 % bei FDA-Warnbriefen, die Lücken in der Marktüberwachung anführten. Automatisierung verändert den Dienstleistungsmix: Veevas Vault RIM reduziert routinemäßige Checklistenarbeit, befreit Autoren für hochwertige narrative Aufgaben und steigert die Nachfrage nach fortgeschrittenen klinischen Bewertungsberichten. Der Markt für Medical Device Regulatory Affairs für Produktregistrierung skaliert nun direkt mit der Einreichungsgeschwindigkeit und nicht mehr ausschließlich mit der Mitarbeiterzahl, was Anbieter dazu ermutigt, Cloud-Tools einzusetzen, um Margen zu erhalten.
Eine Verschiebung zweiter Ordnung betrifft die Vermischung von Schulungs-, Audit- und Marktüberwachungsleistungen. Die Leitlinien der FDA zu Real-World-Evidence aus dem Jahr 2025 öffneten Türen für hybride Datenmodelle, die randomisierte Studien mit Beobachtungsdatensätzen kombinieren und spezialisierte Biostatistik erfordern. Anbieter, die RIM-Automatisierung mit statistischen Dienstleistungen verbinden, sind gut positioniert, um überdurchschnittliche Leistungen zu erzielen, auch wenn die Dokumentationsvolumina weiter steigen. Insgesamt sichern diese Kräfte einen dauerhaften Vorteil für Beratungsunternehmen, die in Workflow-Technologien, KI-gestützte Schreibhilfen und global akkreditierte Qualitätssysteme reinvestieren.



Nach Typ: Diagnostische Geräte übertreffen das Wachstum therapeutischer Geräte
Therapeutische Geräte hielten 42,56 % des Umsatzes im Jahr 2025, angetrieben durch hochwertige Herzimplantate, orthopädische Implantate und Insulinabgabeimplantate, die umfangreiche Vorabmarktstudien und damit große regulatorische Budgets erfordern. Diagnoseplattformen, insbesondere In-vitro-Diagnostika und algorithmische Entscheidungsunterstützungstools, expandieren mit einer schnelleren CAGR von 12,87 %, da Entwicklungszyklen kürzer und Evidenzschwellen niedriger sind. Der Rückstand Europas bei der In-vitro-Diagnostika-Verordnung erzwang bis 2025 weitreichende Neuzertifizierungen und steigerte die Beratungseinnahmen für Testkithersteller erheblich. Die FDA genehmigte im Jahr 2025 89 De-Novo-Diagnosealgorithmen, doppelt so viele wie 2023, was auf eine nachhaltige Pipeline hindeutet, die Schreibaufträge vervielfacht. Der Markt für Medical Device Regulatory Affairs für diagnostische Einreichungen zeigt ein günstiges Gleichgewicht zwischen Volumen und Komplexität, was das Segment für mittelgroße Beratungsunternehmen mit KI-gestützten Vorlagenbibliotheken attraktiv macht.
Therapeutische Einreichungen bleiben arbeitsintensiv, insbesondere nachdem die FDA hybride Evidenzmodelle eingeführt hat, die die Koordination sowohl randomisierter als auch realer Datensätze erfordern. Japans demografisches Profil intensivierte die Nachfrage nach therapeutischen Zulassungen, was sich in einem Anstieg der PMDA-Einreichungen um 12 % im Geschäftsjahr 2024 widerspiegelt. Dennoch behalten Diagnostika den numerischen Vorteil, da jede Plattform typischerweise mehrere Software-Updates hervorbringt, die regulatorische Ergänzungen erfordern. Anbieter, die schnelle Sprints für iterative Diagnoseveröffentlichungen beherrschen, werden daher Marktanteile im breiteren Markt für Medical Device Regulatory Affairs gewinnen.
Nach Dienstleistungsanbieter: Outsourcing gewinnt an Bedeutung, da die Compliance-Last steigt
Ausgelagerte Anbieter sammelten 58,65 % des Umsatzes im Jahr 2025 und bestätigten damit die Präferenz der Hersteller für elastische Kostenstrukturen angesichts unvorhersehbarer Arbeitsspitzen. IQVIAs Segment Technologie- und Analyselösungen erzielte im Jahr 2025 15,4 Milliarden USD und wuchs organisch um 8,2 %, angetrieben durch Cybersicherheits- und EUDAMED-Engagements. LabCorps Covance-Sparte eröffnete im November 2025 ein Zentrum mit 200 Mitarbeitern in Hyderabad, um Indiens niedrige Arbeitskosten für das Segment Produktregistrierung mit einer CAGR von 12,76 % zu nutzen. Dennoch wird erwartet, dass interne Teams jährlich um 13,54 % wachsen, da Konzerne wie Medtronic Integrationsprojekte internalisieren, um proprietäre Datensätze zu schützen. Das Gleichgewicht der Marktanteile für Medical Device Regulatory Affairs wird sich daher zu einem Zwei-Spur-System entwickeln: Ausgelagerte Partner dominieren routinemäßige Einreichungen und ressourcenintensive Spitzen, während interne Abteilungen die Verantwortung für strategische, IP-sensible Dossiers übernehmen.
Kleinere Innovatoren, denen die Größe fehlt, um dediziertes Personal zu rechtfertigen, werden weiterhin auf Berater zurückgreifen, die Pay-as-you-go-Zugang zu KI-gestützten Vorlagen und multinationaler Expertise bieten. Freyr und Promedica verkörpern dieses Modell, indem sie RIM-Technologie lizenzieren, um Zeitpläne zu verkürzen und größere Auftragsforschungsorganisationen beim Preis zu unterbieten, ohne die Qualität zu beeinträchtigen. Die strategische Zweiteilung legt nahe, dass ausgelagerte Spezialisten den Mehrheitsanteil behalten werden, der Umsatzmix jedoch diejenigen begünstigen wird, die bereit sind, in Automatisierung zu reinvestieren.
Geografische Analyse
Nordamerika generierte im Jahr 2025 42,48 % der globalen Ausgaben, gestützt durch die 4.143 genehmigten 510(k)-Einreichungen der FDA und ein unvergleichliches Ökosystem von Auftragsforschungsorganisationen. Durchschnittliche US-amerikanische Prüfzyklen verlängerten sich nach Inkrafttreten der Cybersicherheitsbestimmungen im März 2024 auf 147 Tage, eine Verzögerung, die die mit Abhilfemaßnahmen und Wiedereinreichungen verbundenen Beratungsstunden vergrößerte. Health Canada richtete im Juni 2025 die Leitlinien für Software als Medizinprodukt an den US-amerikanischen Erwartungen aus, was eine grenzüberschreitende Dossierharmonisierung erforderte und bilaterale Beratungsengagements ankurbelte. Mexiko verkürzte seine Klasse-II-Genehmigungen im September 2024 auf sieben Monate, was Unternehmen dazu ermutigte, Compliance-Workflows innerhalb eines nordamerikanischen Knotenpunkts zu regionalisieren und den gesamten adressierbaren Ausgabenrahmen im Markt für Medical Device Regulatory Affairs zu vergrößern.
Asien-Pazifik ist die am schnellsten wachsende Region mit einer CAGR von 11,54 % bis 2031, angetrieben durch weitreichende Reformen in China, Indien, Japan und Südkorea. Chinas neunmonatige Prioritätsprüfung für innovative Klasse-III-Geräte verlagert das Budget von klinischen Verzögerungen hin zu vorgelagerten regulatorischen Strategien und steigert die Nachfrage nach Beratern in Peking und Shanghai. Indiens Regelreform von 2024 reduzierte die Dokumentation um 30 %, aber die Notwendigkeit, lokalisierte Vorlagen zu interpretieren, macht ausländische Hersteller weiterhin von lokalen Beratungsunternehmen abhängig. Japans Behörde für pharmazeutische und medizinische Geräte bearbeitete im Geschäftsjahr 2024 1.247 Anträge, da die alternde Bevölkerung die Nachfrage nach therapeutischen Geräten ankurbelt. Südkoreas gegenseitiges Anerkennungsabkommen mit der FDA vom März 2025 senkte die durchschnittlichen Kosten für doppelte Einreichungen um 20 % und lenkte zusätzliche Projekte zu Beratern in Seoul.
Europa blieb im Jahr 2025 ein starker Ausgabenmarkt aufgrund der schrittweisen Einführung der Medizinprodukteverordnung, der In-vitro-Diagnostika-Verordnung und des EUDAMED-Starts im Mai 2026. Deutschland, das Vereinigte Königreich, Frankreich, Italien und Spanien machten mehr als 60 % der regionalen Beratungsausgaben aus, da lokale Benannte-Stellen-Warteschlangen die Abhängigkeit von externen Dossierautoren erhöhten. Brasiliens 30-prozentige Gebührensenkung für Kleinunternehmen und seine Anerkennung von FDA-Zulassungen belebten die südamerikanischen Ausgaben, während Saudi-Arabien und die Vereinigten Arabischen Emirate die Harmonisierung des Golfkooperationsrats für Gesundheit vorantrieben und die Compliance im Nahen Osten unter einheitlichen Vorlagen konsolidierten. Insgesamt erweitern diese Maßnahmen den geografischen Fußabdruck des Marktes für Medical Device Regulatory Affairs, ohne die Vorrangstellung Nordamerikas und Europas beim Gesamtvolumen zu schmälern.

Wettbewerbslandschaft
Der Sektor bleibt moderat fragmentiert; die 10 bedeutendsten Anbieter halten etwa 35 % des Umsatzes im Jahr 2025. IQVIA, ICON und LabCorp führen das ausgelagerte Segment an und nutzen End-to-End-Angebote, die regulatorische Beratung, klinische Studienüberwachung und Marktüberwachung umfassen. IQVIAs organisches Wachstum von 8,2 % im Jahr 2025 unterstreicht den Magneteffekt von Komplettlösungen für Cybersicherheitsdokumentation und EUDAMED-Uploads. Intertek und SGS diversifizierten sich in den Jahren 2024–2025 von der Prüfung in die regulatorische Beratung und vermarkteten gebündelte Audit- und Einreichungspakete, die mittelgroße Hersteller ansprechen, die Einfachheit suchen. Freyr, Promedica und andere Nischenspezialisten setzen KI-gestützte Vorlagen ein, um Fertigstellungszeiten zu verkürzen und Verträge im mittleren Segment zu gewinnen.
Chancen in weißen Flecken liegen in KI-gestützter regulatorischer Informationsgewinnung und der Lokalisierung für Schwellenmärkte. Veeva bearbeitete 12.000 Einreichungen über Vault RIM und demonstrierte damit, dass Automatisierung Skalenvorteile ausgleicht und Kommoditisierung verhindert. Talentknappheit ist die begrenzende Variable; das Mitgliederwachstum der Gesellschaft für Fachleute im Bereich Regulatory Affairs von 3,1 % bleibt hinter der Nachfrage zurück und zwingt Unternehmen, Offshore-Hubs und robuste interne Schulungsakademien einzurichten. Akquisitionspipelines bleiben aktiv, da größere Auftragsforschungsorganisationen technologiereiche Boutiquen sondieren, um Expertise-Lücken zu schließen, wie SGSs Mehrheitsbeteiligung an einem Beratungsunternehmen in Bangalore und Medtronics Plattformkonsolidierung zeigen.
Marktführer im Bereich Medical Device Regulatory Affairs
ICON, Plc
IQVIA, Inc.
Laboratory Corporation of America Holdings
Integer Holdings Corporation
SGS Société Générale de Surveillance SA
- *Haftungsausschluss: Hauptakteure in keiner bestimmten Reihenfolge sortiert

Jüngste Branchenentwicklungen
- August 2025: Die britische Behörde für Arzneimittel und Gesundheitsprodukte (MHRA) startete einen neuen Frühzugangsdienst zur Unterstützung der schnelleren Einführung innovativer Medizinprodukte, insbesondere in Bereichen mit ungedecktem klinischen Bedarf.
- Juni 2024: IMed Consultancy hat ein Weißbuch veröffentlicht, das die regulatorische Landschaft für KI/ML-Medizinprodukte in den USA, dem Vereinigten Königreich und der EU detailliert beschreibt und einzigartige Herausforderungen hervorhebt, wie die Hochrisikoeinstufung des EU-KI-Gesetzes, die mit der Medizinprodukteverordnung/In-vitro-Diagnostika-Verordnung für selbstlernende Systeme in Konflikt steht, die wachsenden Genehmigungen der FDA und den sich entwickelnden Rahmen des Vereinigten Königreichs nach dem Brexit, wobei die Schlüsselthemen auf Sicherheit, Transparenz und Lebenszyklusmanagement für diese dynamischen Technologien ausgerichtet sind.


Umfang des globalen Berichts zum Markt für Medical Device Regulatory Affairs
Gemäß dem Umfang des Berichts umfasst Medical Device Regulatory Affairs die Sicherstellung, dass Medizinprodukte nationalen und internationalen Vorschriften und Standards entsprechen. Es umfasst die Verwaltung von Genehmigungen, Registrierungen und Dokumentationen, die für den Marktzugang erforderlich sind. Dieses Feld stellt die Sicherheit, Wirksamkeit und rechtliche Konformität von Medizinprodukten während ihres gesamten Lebenszyklus sicher.
Der Markt für Medical Device Regulatory Affairs ist segmentiert nach Dienstleistungen (Regulierungsberatung, Rechtliche Vertretung, Regulatorisches Schreiben und Publizieren, Produktregistrierung und klinische Studienanträge sowie sonstige Dienstleistungen), Typ (Diagnostisch und Therapeutisch), Dienstleistungsanbieter (Intern und Ausgelagert) sowie Geografie (Nordamerika, Europa, Asien-Pazifik, Naher Osten und Afrika sowie Südamerika). Der Marktbericht umfasst auch die geschätzten Marktgrößen und Trends für 17 Länder in den wichtigsten Regionen weltweit. Der Bericht bietet den Wert (in Millionen USD) für die oben genannten Segmente.
| Regulierungsberatung |
| Rechtliche Vertretung |
| Regulatorisches Schreiben und Publizieren |
| Produktregistrierung und klinische Studienanträge |
| Sonstige Dienstleistungen |
| Diagnostisch |
| Therapeutisch |
| Intern |
| Ausgelagert |
| Nordamerika | Vereinigte Staaten |
| Kanada | |
| Mexiko | |
| Europa | Deutschland |
| Vereinigtes Königreich | |
| Frankreich | |
| Italien | |
| Spanien | |
| Übriges Europa | |
| Asien-Pazifik | China |
| Japan | |
| Indien | |
| Australien | |
| Südkorea | |
| Übriges Asien-Pazifik | |
| Naher Osten und Afrika | Golfkooperationsrat |
| Südafrika | |
| Übriger Naher Osten und Afrika | |
| Südamerika | Brasilien |
| Argentinien | |
| Übriges Südamerika |
| Nach Dienstleistungen | Regulierungsberatung | |
| Rechtliche Vertretung | ||
| Regulatorisches Schreiben und Publizieren | ||
| Produktregistrierung und klinische Studienanträge | ||
| Sonstige Dienstleistungen | ||
| Nach Typ | Diagnostisch | |
| Therapeutisch | ||
| Nach Dienstleistungsanbieter | Intern | |
| Ausgelagert | ||
| Geografie | Nordamerika | Vereinigte Staaten |
| Kanada | ||
| Mexiko | ||
| Europa | Deutschland | |
| Vereinigtes Königreich | ||
| Frankreich | ||
| Italien | ||
| Spanien | ||
| Übriges Europa | ||
| Asien-Pazifik | China | |
| Japan | ||
| Indien | ||
| Australien | ||
| Südkorea | ||
| Übriges Asien-Pazifik | ||
| Naher Osten und Afrika | Golfkooperationsrat | |
| Südafrika | ||
| Übriger Naher Osten und Afrika | ||
| Südamerika | Brasilien | |
| Argentinien | ||
| Übriges Südamerika | ||


Im Bericht beantwortete Schlüsselfragen
Welchen prognostizierten Wert wird der Markt für Medical Device Regulatory Affairs bis 2031 erreichen?
Der Markt wird voraussichtlich bis 2031 14,16 Milliarden USD erreichen.
Welches Dienstleistungssegment wird bis 2031 am schnellsten wachsen?
Produktregistrierung und klinische Studienanträge, mit einer Expansion von 12,76 % CAGR.
Welche Region wächst bei den Ausgaben für Regulatory Affairs am schnellsten?
Asien-Pazifik, mit einer Wachstumsrate von 11,54 % CAGR.
Welchen Anteil hielten ausgelagerte Anbieter im Jahr 2025?
Ausgelagerte Anbieter sicherten sich 58,65 % des Umsatzes im Jahr 2025.
Wie wirken sich Cybersicherheitsmandate auf den Umfang der Einreichungen aus?
Neue Vorschriften fügen typischen 510(k)-Einreichungen 40–60 Seiten hinzu und verlängern die Prüffristen um bis zu vier Wochen.
Welche Gerätekategorie wächst schneller, Diagnostik oder Therapeutik?
Diagnoseplattformen wachsen schneller mit einer CAGR von 12,87 % gegenüber therapeutischen Geräten.
Seite zuletzt aktualisiert am: